Для расчетов
тепловых эффектов используют величину,
называемую энтальпией.
Энтальпией
(теплотой) образования химического
соединения ∆ HT
называется изменение энтальпии
в процессе получения 1 моля этого
соединения из простых веществ (элементов)
при постоянном давлении.
Так, например,
энтальпией образования этилового спирта
C2H5OHназывается изменение энтальпии при
реакции углерода, кислорода и водорода
при постоянном давлении
2C
+ 3H2 + 1/2
O2→ CH3
– CH2 – OH
И наоборот, реакции
CH2
= CH2+H2O→CH3
– CH2
– OH
CH3
– CHO+H2→CH3
– CH2
– OH
не являются
реакциями ”образования” спирта, хотя
и приводят к синтезу этилового спирта.
Важным термодинамическим
представлением является понятие о
стандартном состоянии вещества, под
которым понимается его реальное состояние
при 298 Ки атмосферном давлении (илиp = 101 кПа).
Стандартной
энтальпией (теплотой) образования
химического соединения
![]() называется изменение энтальпии в
называется изменение энтальпии в
процессе образования 1 моля этого
соединения, находящегося в стандартном
состоянии, из простых веществ, также
находящихся в стандартных состояниях
и термодинамически устойчивых при
данной температуре фазах и модификациях.
Стандартные
энтальпии образования простых веществ
принимают равными нулю, если их агрегатные
состояния и модификации устойчивы при
стандартных условиях.
Стандартная
энтальпия образования соединения –
мера его термодинамической устойчивости,
прочности, количественное выражение
энергетических свойств соединения.
Если стандартная энтальпия образования
отрицательна, то соединение более
устойчиво, чем элементы, из которых оно
состоит, или наоборот, если она
положительна, то соединение менее
устойчиво. Из различных молекул более
устойчивы те, энтальпии, образования
которых меньше.
Например, этан (∆
H298 = –
24,82 ккал/моль)более устойчив, чем
этилен
![]()
Термохимические
расчеты.Значения энтальпий
образования позволяет определить
изменения энтальпии, сопровождающие
реакции, не прибегая к прямым измерениям.
В основе большинства
термохимических расчетов лежит следствие
из закона Гесса:
Тепловой эффект
химической реакции равен сумме теплот
(энтальпий) образования продуктов
реакции за вычетом суммы теплот
(энтальпий) образования исходных веществ.
Так, для уравнения
вида
aA
+ bB +…= dD
+ eE +…
Тепловой эффект
∆ Hопределяется
равенством
![]() =
=
![]()
При этом следует
иметь в виду, что при алгебраическом
суммировании следует учитывать
стехиометрические коэффициенты в
уравнении реакции (а, в, d,
e).
Приведенное
уравнение позволяет определять как
тепловой эффект реакции по известным
энтальпиям образования веществ,
участвующих в реакции, так и одну из
энтальпий образования, если известны
тепловой эффект реакции и все остальные
энтальпии образования.
В настоящее время
энтальпии образования известны примерно
для четырех тысяч веществ, и это позволяет
расчётным путем установить тепловые
эффекты самых разнообразных реакций.
Пример 1. Расчет
теплового эффекта реакции по стандартным
энтальпиям образования веществ.
Al2O3
(к)
+3SO3 (г)
= Al2(SO4)3
(к)
![]() =-1675
=-1675
кДж/моль;
![]() =-395,
=-395,
2 кДж/моль
![]() =-3434
=-3434
кДж/моль
Тепловой эффект
реакции
![]() определяется по уравнению:
определяется по уравнению:
![]()
После подстановки
значений стандартных энтальпий
![]() -3434-(-1675-3.395,
-3434-(-1675-3.395,
2)=-599 кДж/моль
Видно, что
![]() <
<
0, т.е. это реакция экзотермическая.
Пример 2. Расчет
энтальпии образования химического
соединения по известному тепловому
эффекту реакции.
Определить
![]() в реакции:
в реакции:
CH4
(г) + 2 H2O
(г) = CO2
(г) + 4 H2
(г)
![]() =-241,82
=-241,82
кДж/моль;
![]() =-393,51
=-393,51
кДж/моль;
![]() =
=
+164,98 кДж
Стандартный
тепловой эффект реакции равен:
![]() ,
,
откуда
![]()
После подстановки
стандартных энтальпий образования
веществ:
![]() (-1
(-1
моль∙ 393,51 кДж/моль + 2 моль ∙ – 241,82
кДж/моль – 164,98 кДж) / 1 моль = – 74,85 кДж/моль
Видно, что реакция
образования метана является экзотермической.
Пример 3. Расчет
теплового эффекта реакции, который
экспериментально определить невозможно.
Реакция 1) C
+ O2 = CO2,
![]()
= -396 кДж/моль
может быть также
осуществлена в две стадии
2)
C + 1/2 O2
= CO,
![]()
= -110, 5 кДж/моль
3)
CO + 1/2 O2
= CO2
Тепловой эффект
реакции 2) можно провести экспериментально
и определить
![]() =-110, 5 кДж/моль.
=-110, 5 кДж/моль.
А вот тепловой
эффект реакции 3) экспериментально
осуществить и определить невозможно,
поэтому
![]() реакции
реакции
3) получают расчетным путем, используя
закон Гесса
![]()
![]()
![]() – 396 – (-110, 5) = – 285,5
– 396 – (-110, 5) = – 285,5
кДж/моль
С помощью
термохимических расчетов можно определить
энергию химических связей, энергии
кристаллических решеток, теплоты
растворения и гидратации, тепловые
эффекты фазовых превращений и т.д.
Энтальпия есть
возрастающая функция температуры,
непрерывная во всей области изменения
последней, когда существует данная
фаза. Для расчета теплового эффекта при
температурах, более высоких, чем
стандартные, используют уравнение
Кирхгофа:
![]()
где
![]() – стандартный тепловой эффект реакции
– стандартный тепловой эффект реакции
приT = 298 К;
![]() –
–
стандартный тепловой эффект реакции
при T.
![]() разность молярных
разность молярных
изобарных теплоемкостей всех продуктов
реакции и молярных изобарных теплоемкостей
всех исходных веществ, т.е.
![]()
Расчеты по уравнению
Кирхгофа показывают, что в том интервале
температур, который может иметь
практическое значение, изменение
величины теплового эффекта реакции
невелико.
Так, например,
тепловой эффект процесса 1/2 N2
+ 1/2 O2
= NO (г)при
повышении температуры от298до4000Кизменяется всего лишь на2,0
кДж/моль. Еще меньше влияние давления
на тепловой эффект реакции. Так, для
реакции синтеза аммиака из азота и
водорода различие между величинами
![]() приp = 101 кПаиp
приp = 101 кПаиp
= 50 МПане превышает5 %.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
![]()
Загрузить PDF
![]()
Загрузить PDF
Во время химических реакций происходит поглощение или выделение тепла в окружающую среду. Такой теплообмен между химической реакцией и окружающей средой называется энтальпией, или H. Однако измерить энтальпию напрямую невозможно, поэтому принято рассчитывать изменение температуры окружающей среды (обозначаемое ∆H). ∆H показывает, что в ходе химической реакции происходит выделение тепла в окружающую среду (экзотермическая реакция) или поглощение тепла (эндотермическая реакция). Рассчитывается энтальпия так: ∆H = m x s x ∆T, где m — масса реагентов, s — теплоемкость продукта реакции, ∆T — изменение температуры в результате реакции.
-

1
Определите реагенты и продукты реакции. Любая химическая реакция имеет реагенты и продукты реакции. Продукт реакции создается в результате взаимодействия реагентов. Другими словами реагенты — это ингредиенты в рецепте, а продукт реакции — это готовое блюдо. Чтобы найти ∆H реакции, необходимо знать реагенты и продукты реакции.
- Например, необходимо найти энтальпию реакции образования воды из водорода и кислорода: 2H2 (водород) + O2 (кислород) → 2H2O (вода). В этой реакции H2 и O2 – реагенты, а H2O — продукт реакции.
-
2
Определите общую массу реагентов. Далее необходимо подсчитать массу реагентов. Если вы не можете взвесить их, то подсчитайте молекулярную массу, чтобы найти фактическую. Молекулярная масса — это постоянная, которую можно найти в периодической таблице Менделеева или в других таблицах молекул и соединений. Умножьте массу каждого реагента на число молей.
- В нашем примере реагенты водород и кислород имеют молекулярные массы 2 г и 32 г соответственно. Поскольку мы используем 2 моль водорода (коэффициент в химической реакции перед водородом H2) и 1 моль кислорода (отсутствие коэффициента перед O2 обозначает 1 моль), то общая масса реагентов рассчитывается следующим образом:
2 × (2 г) + 1 × (32 г) = 4 г + 32 г = 36 г
- В нашем примере реагенты водород и кислород имеют молекулярные массы 2 г и 32 г соответственно. Поскольку мы используем 2 моль водорода (коэффициент в химической реакции перед водородом H2) и 1 моль кислорода (отсутствие коэффициента перед O2 обозначает 1 моль), то общая масса реагентов рассчитывается следующим образом:
-
3
Определите теплоемкость продукта. Далее определите теплоемкость продукта реакции. Каждая молекула имеет определенную величину теплоемкости, которая является постоянной. Найдите эту постоянную в таблицах учебника по химии. Существует несколько единиц измерения теплоемкости; в наших расчетах мы будем использовать Дж/г°C.
- Обратите внимание на то, что при наличии нескольких продуктов реакции вам потребуется рассчитать теплоемкость каждого, а затем сложить их, чтоб получить энтальпию всей реакции.
- В нашем примере, продукт реакции — вода, которая имеет теплоемкость 4,2 Дж/г°C.
-
4
Найдите изменение температуры. Теперь мы найдем ∆T — разницу температур до и после реакции. Из начальной температуры (T1) вычтите конечную температуру (T2). Чаще всего в задачах по химии используется шкала Кельвина (К) (хотя по шкале Цельсия (°С) получится тот же результат).
- В нашем примере давайте предположим, что начальная температура реакции была 185 K, а после реакции стала 95 K, значит, ∆T вычисляется так:
∆T = T2 – T1 = 95 K – 185 K = -90 K
- В нашем примере давайте предположим, что начальная температура реакции была 185 K, а после реакции стала 95 K, значит, ∆T вычисляется так:
-
5
Найдите энтальпию по формуле ∆H = m x s x ∆T. Если известна m — масса реагентов, s — теплоемкость продукта реакции и ∆T — изменение температуры, то можно подсчитать энтальпию реакции. Подставьте значения в формулу ∆H = m x s x ∆T и получите энтальпию. Результат вычисляется в Джоулях (Дж).
- В нашем примере энтальпия вычисляется так:
∆H = (36 г) × (4,2 ДжK – 1 г – 1) × (-90 K) = -13608 Дж
- В нашем примере энтальпия вычисляется так:
-
6
Определите, выделяется или поглощается энергия в ходе рассматриваемой реакции. Одна из самых распространенных причин, по которой требуется вычислить ∆H на практике, — узнать, будет ли реакция экзотермической (выделение тепла и снижение собственной энергии) или эндотермической (поглощение тепла из окружающей среды и повышение собственной энергии). Если значение ∆H положительное, значит, реакция эндотермическая. Если отрицательное, значит, реакция экзотермическая. Чем больше абсолютное значение ∆H, тем больше энергии выделяется или поглощается. Будьте осторожны, если собираетесь проводить практический опыт: во время реакций с высоким значением энтальпии может произойти большое высвобождение энергии, и если оно протекает быстро, то может привести ко взрыву.
- В нашем примере конечный результат получился равным -13608 Дж. Перед значением энтальпии отрицательный знак, а это означает, что реакция экзотермическая. Горячие газы (в виде пара) H2 и O2 должны выделить некоторое количество тепла, чтобы образовать молекулу воды, то есть реакция образования H2O является экзотермической.
Реклама






-
1
Подсчитайте энергию связей для оценки энтальпии. Почти все химические реакции приводят к разрыву одних связей и образованию других. Энергия в результате реакции не возникает ниоткуда и не разрушается: это та энергия, которая требуется для разрыва или образования этих связей. Поэтому изменение энтальпии всей реакции можно довольно точно оценить путем суммирования энергии этих связей.
- Например, рассмотрим реакцию: H2 + F2 → 2HF. В этом случае, энергия для разрыва связи в молекуле H2 составляет 436 кДж/моль, а энергия для разрыва F2 составляет 158 кДж/моль.[1]
Наконец, энергия необходимая для образования связи в молекуле HF из H и F = -568 кДж/моль.[2]
Умножаем последнее значение на 2, так как в ходе реакции получаем 2 моль HF: 2 × -568 = -1136 кДж/моль. Складываем значения:
436 + 158 + (-1136) = -542 кДж/моль.
- Например, рассмотрим реакцию: H2 + F2 → 2HF. В этом случае, энергия для разрыва связи в молекуле H2 составляет 436 кДж/моль, а энергия для разрыва F2 составляет 158 кДж/моль.[1]
-
2
Используйте энтальпию образования для оценки энтальпии. Энтальпия образования позволяет рассчитать ∆H через вычисление реакций образования реагентов и продуктов. Если известна энтальпия образования продуктов реакции и реагентов, то вы можете оценить энтальпию в целом путем сложения, как и в случае энергии, рассмотренном выше.
- Например, рассмотрим следующую реакцию: C2H5OH + 3O2 → 2CO2 + 3H2O. Мы знаем, что энтальпия образования рассчитывается:[3]
C2H5OH → 2C + 3H2 + 0,5O2 = 228 кДж/моль
2C + 2O2 → 2CO2 = -394 × 2 = -788 кДж/моль
3H2 + 1.5 O2 → 3H2O = -286 × 3 = -858 кДж/моль
Теперь необходимо сложить значения образованных веществ, полученные выше, для определения энтальпии реакции: C2H5OH + 3O2 → 2CO2 + 3H2O,
228 + -788 + -858 = -1418 кДж/моль.
- Например, рассмотрим следующую реакцию: C2H5OH + 3O2 → 2CO2 + 3H2O. Мы знаем, что энтальпия образования рассчитывается:[3]
-
3
Не забывайте о знаках перед значениями энтальпии. При вычислении энтальпии образования формулу для определения энтальпии реакции продукта вы переворачиваете, и знак энтальпии должен поменяться. Другими словами, если вы переворачиваете формулу, то знак энтальпии должен смениться на противоположный.
- В примере обратите внимание на то, что реакция образования для продукта C2H5OH записана наоборот. C2H5OH → 2C + 3H2 + 0,5O2 то есть C2H5OH распадается, а не синтезируется. Поэтому знак перед энтальпией в такой реакции положительный, 228 кДж/моль, хотя энтальпия образования C2H5OH составляет -228 кДж/моль.
Реклама



-
1
Возьмите чистую емкость и налейте туда воды. Увидеть принципы энтальпии в действии нетрудно — достаточно провести простой опыт. Необходимо, чтобы на результат эксперимента не повлияли посторонние загрязнители, так что емкость нужно вымыть и простерилизовать. Ученые для измерения энтальпии используют специальный закрытые контейнеры — калориметры, но вам вполне подойдет стеклянный стакан или колба. Заполните емкость чистой водопроводной водой комнатной температуры. Желательно проводить эксперимент в прохладном помещении.
- Для эксперимента желательно использовать небольшую емкость. Мы будем рассматривать энтальпию реакции воды с «Алка-Зельтцер», поэтому, чем меньше воды используется, тем более очевидным будет изменение температуры.
-
2
Поместите термометр в емкость. Возьмите термометр и опустите его в емкость с водой так, чтобы граница прочтения температуры была ниже уровня воды. Снимите показания термометра — это будет начальная температура, T1.
- Предположим, что температуры воды 10 °C. Мы будем использовать это значение для демонстрации принципов энтальпии.
-
3
Добавьте в емкость одну таблетку «Алка-Зельтцер». Готовы начать опыт? Бросьте в воду одну таблетку «Алка-Зельтцер». Она сразу начнет пузыриться и шипеть. Это происходит из-за реакции между бикарбонатом (HCO3–) и лимонной кислотой (H+). В результате образуются вода и углекислый газ по формуле: 3HCO3− + 3H+ → 3H2O + 3CO2.
-
4
Измерьте конечную температуру. Наблюдайте за ходом реакции: таблетка «Алка-Зельтцер» будет постепенно растворяться. Когда она растворится полностью, измерьте температуру еще раз. Вода должна стать немного холоднее. Если температура воды напротив стала выше начальной, значит, эксперименту помешали какие-то внешние факторы (например, слишком теплое помещение, где проводился эксперимент).
- Предположим, что температура составляет теперь 8 °C.
-
5
Подсчитаем энтальпию реакции. Когда таблетка «Алка-Зельтцер» вступает в реакцию с водой, образуются вода и углекислый газ (те самые шипучие пузырьки) и происходит снижение температуры (это тот результат, который должен получиться, если опыт прошел успешно). Можно сделать вывод, что данная химическая реакция является эндотермической, то есть она сопровождается поглощением энергии из окружающей среды — в данном случае из воды. В результате температура воды снижается.
- В нашем эксперименте температура воды снизилась на два градуса. Это согласуется с теорией: реакция растворения «Алка-Зельтцер» в воде эндотермическая и сопровождается небольшим поглощением энергии.
Реклама





Советы
- В подсчетах используется шкала Кельвина (K) — это температурная шкала, аналогическая шкале Цельсия, и часто применяемая в химии и физике. Чтобы перевести значение градусов Цельсия в кельвины, необходимо добавить или вычесть 273 градуса: K = °C + 273.
Реклама
Об этой статье
Эту страницу просматривали 115 669 раз.
Была ли эта статья полезной?
Материалы из методички: Сборник задач по теоретическим основам химии для студентов заочно-дистанционного отделения / Барботина Н.Н., К.К. Власенко, Щербаков В.В. – М.: РХТУ им. Д.И. Менделеева, 2007. -155 с.
Тепловой эффект процесса
Термохимические уравнения
Закон Гесса
Следствия из закона Гесса
Стандартные термодинамические величины
Стандартные энтальпии образования и сгорания
Примеры решения задач
Задачи для самостоятельного решения
Тепловой эффект процесса
Количество выделенной (или поглощенной) теплоты Q в данном процессе называют тепловым эффектом процесса. Экзотермической является реакция, протекающая с выделением теплоты, а эндотермической – с поглощением теплоты из окружающей среды.
Для лабораторных и промышленных процессов наиболее типичен изобарный режим (Р=const). Поэтому обычно рассматривают тепловой эффект при Р,Т = const, т.е. изменение энтальпии процесса ΔН.
Следует отметить, что абсолютные значения энтальпии Н определить не представляется возможным, так как не известна абсолютная величина внутренней энергии.
Для экзотермической реакции (Q > 0) ΔН < 0, а в эндотермическом процессе (Q < 0) ΔН > 0.
Термохимические уравнения
Химические уравнения, в которых дополнительно указывается величина изменения энтальпии реакции, а также агрегатное состояние веществ и температура, называются термохимическими уравнениями.
В термохимических уравнениях отмечают фазовое состояние и аллотропные модификации реагентов и образующихся веществ: г – газообразное, ж – жидкое, к – кристаллическое; S(ромб), S(монокл), С(графит), С(алмаз) и т.д.
Важно подчеркнуть, что с термохимическими уравнениями можно проводить алгебраические операции сложения, вычитания, деления, умножения.
Закон Гесса
Изменение энтальпии (внутренней энергии) химической реакции зависит от вида, состояния и количества исходных веществ и продуктов реакции, но не зависит от пути процесса.
Следствия из закона Гесса
- Изменение энтальпии реакции равно сумме энтальпий образования продуктов реакции за вычетом суммы энтальпий образования исходных веществ (суммирование проводится с учетом стехиометрических коэффициентов).
- Изменение энтальпии реакции равно сумме энтальпий сгорания исходных веществ за вычетом суммы энтальпий сгорания продуктов реакции (суммирование проводится с учетом стехиометрических коэффициентов).
Стандартные термодинамические величины
Стандартные термодинамические величины – это такие величины, которые относятся к процессам, все ингредиенты которых находятся в стандартных состояниях.
Стандартным состоянием вещества, находящегося в конденсированной фазе (кристаллической или жидкой), является реальное состояние вещества, находящегося при данной температуре и давлении 1 атм.
Следует подчеркнуть, что стандартное состояние может иметь место при любой температуре.
Обычно тепловой эффект (изменение энтальпии) реакции приводится для температуры 25оС (298,15 К) и давления 101,325 кПа (1 атм), т.е. указывается стандартная энтальпия ΔНо298.
Стандартные энтальпии образования и сгорания
Стандартная энтальпия образования ΔНоf,298 (или ΔНообр,298) – это изменение энтальпии в процессе образования данного вещества (обычно 1 моль), находящегося в стандартном состоянии, из простых веществ, также находящихся в стандартном состоянии, причем простые вещества присутствуют в наиболее термодинамически устойчивых состояниях при данной температуре.
Например, ΔНof,298(Н2О(ж)) = — 285,83 кДж/моль соответствует изменению энтальпии в процессе
Н2(г) + ½O2(г) = Н2О(ж)
при Т = 298,15 К и Р = 1 атм.
Стандартная энтальпия образования простых веществ равна нулю по определению (для наиболее устойчивых их модификаций при данной температуре).
Стандартной энтальпией сгорания ΔНoсгор,298 называют энтальпию сгорания вещества (обычно 1 моль), находящегося в стандартном состоянии с образованием СО2(г), Н2О(ж) и других веществ, состав которых должен быть специально указан. Все продукты сгорания также должны находиться в стандартном состоянии.
Примеры решения задач
Задача 1. Используя справочные термодинамические данные вычислить ΔНo298 реакции:
2H2S(г) + 3O2(г) = 2SO2(г) + 2H2O(ж); ΔНoх.р.,298 = ?
Решение. Решим задачу, используя оба следствия из закона Гесса. Ниже для исходных веществ и продуктов реакции приведены значения энтальпий образования и сгорания в кДж/моль (энтальпия сгорания сероводорода до SO2(г) и H2O(ж)):
| Вещество | H2S(г) | O2(г) | SO2(г) | H2O(ж) |
| ΔНof,298 | -20,60 | 0 | -296,90 | -285,83 |
| ΔНoсгор,298 | -562,10 | 0 | 0 | 0 |
Cогласно первому следствию закона Гесса энтальпия этой реакции ΔНох.р. равна:
ΔНох.р.,298 = 2ΔНоf,298(SO2(г)) + 2ΔНоf,298(H2O(ж)) — 2ΔНоf,298(H2S(г)) — 3ΔНоf,298(O2(г)) = 2(- 296,90) + 2(- 285,83) — 2(- 20,60) = — 1124,21 кДж.
В соответствии со вторым следствием закона Гесса получаем:
ΔНох.р.,298 = 2ΔНосгор,298(H2S(г)) = 2(-562,10) = — 1124,20 кДж.
Задача 2. Вычислите ΔНо298 реакции N2(г) + 3H2(г) = 2NH3(г), используя следующие данные:
4NH3(г) + 3O2(г) = 2N2(г) + 6H2O(ж); ΔНо1 = -1531,22 кДж;
2H2O(ж) = O2(г) + 2H2(г); ΔНо2= 571,66 кДж.
Определите стандартную энтальпию образования NH3(г).
Решение. Поскольку с термохимическими уравнениями можно производить все алгебраические действия, то искомое уравнение получится, если:
-
-
- разделить на два тепловой эффект первого уравнения и изменить его знак на противоположный, т.е:
-
N2(г) + 3H2O(ж) = 2NH3(г) + 3/2O2(г); ΔНо = 765,61 кДж;
-
-
- умножить на 3/2 второе уравнение и соответствующую ему величину δНo , изменив ее знак на противоположный:
-
3/2O2(г) + 3H2(г) = 3H2O(ж); ΔНо = -857,49 кДж;
-
-
- сложить полученные первое и второе уравнения.
-
Таким образом, тепловой эффект реакции N2(г) + 3H2(г) = 2NH3(г) равен:
Δ Но298 = (- ΔНо1/2) + (- 3/2·ΔНо2) = 765,61 + (- 857,49) = — 91,88 кДж.
Поскольку в рассматриваемой реакции образуется 2 моль NH3(г), то
ΔНоf,298(NH3(г)) = — 91,88/2 = — 45,94 кДж/моль.
Задача 3. Определите энтальпию процесса
CuSO4(к) + 5H2O(ж) = CuSO4·5H2O(к)
если при 298,15 К энтальпия растворения CuSO4(к) в n моль Н2О с образованием раствора CuSO4(р-р, nH2O) равна –40, а энтальпия растворения CuSO4·5H2O(к) с образованием раствора той же концентрации равна +10,5 кДж/моль.
Решение. Составляем цикл Гесса:

ΔНо1 = ΔНо2 + ΔНох (по закону Гесса). Отсюда получаем:
ΔНох = ΔНо1 – ΔНо2 = – 40,0 – 10,5 = -50,5 кДж.
Другой вариант решения.
CuSO4(к) + (n H2O) = CuSO4(р-р, n H2O); ΔНо1 = – 40,0 кДж; (1)
CuSO4(к) + 5H2O(ж) = CuSO4·5H2O(к); ΔНох = Но2; (2)
CuSO4·5H2O(к) + (n – 5)H2O = CuSO4(р-р, nH2O); ΔНо3 = 10,5 кДж. (3)
По закону Гесса: ΔНо1 = ΔНох+ ΔНо3, т.е. при сложении уравнений (2) и (3) получим уравнение (1).
Задача 4. Вычислите энтальпию образования химической связи С= С в молекуле этилена, если его стандартная энтальпия образования равна 52,3 кДж/моль, энтальпия возгонки графита составляет 716,7 кДж/моль, энтальпия атомизации водорода равна +436,0 кДж/моль, энтальпия образования связи С–Н равна –414,0 кДж/моль.
Решение. Составляем цикл Гесса:
ΔНоf,298(С2Н4(г)) = 2ΔНовозг(С(графит)) + 2ΔНоатом (H2(г)) + ΔНо(С= С) + 4ΔНо(С–Н).
ΔНо(С = С) = 52,3 — 2·716,7 — 2·436,0 + 4·414,0 = — 597,1 кДж/моль.
Задачи для самостоятельного решения
1. Составьте уравнение реакции, для которой ΔНо соответствует стандартной энтальпии образования ВaCl2·2H2O(к).
2. Определить ΔНо298 реакции:
CH3CНO(ж) + H2(г) = CH3CH2OH(ж),
если ΔНосгор,298(CH3CНO(ж)) = — 1193,07 кДж/моль; ΔНосгор,298(CH3CH2OH(ж)) = — 1370,68 кДж/моль; ΔНоf,298(Н2О(ж)) = — 285,83 кДж/моль.
10.3. Энтальпии растворения BaCl2(к) и BaCl2·2H2O(к) с образованием раствора хлорида бария (с мольным отношением BaCl2: H2O = 1: 500) соответственно равны –11,18 и 18,74 кДж/моль.
Определить величину ΔНо присоединения воды к BaCl2(к) с образованием BaCl2·2H2O(к).
10.4. Рассчитать энтальпию связи в молекуле NO на основании следующих термохимических уравнений:
N2(г) + O2(г) = 2NO(г); ΔНо298 = +182,52 кДж;
2O(г) = O2(г); ΔНо298 = — 498,34 кДж;
N2(г) = 2N(г); ΔНо298 = +945,42 кДж.
10.5. Вычислить ΔНо298 реакции 2C(г) + 2H2(г) = C2H4(г), используя следующие термохимические уравнения:
С(графит) = С(г) ; ΔНо298 = +716,67 кДж;
С2H4(г) + 3O2(г) = 2CO2(г) + 2H2O(г); ΔНо298 = — 1322,94 кДж;
C(графит) + O2(г) = CO2(г); ΔНо298 = — 393,51 кДж;
H2(г) + 1/2O2(г) = H2O(г); ΔНо298 = — 241,81 кДж.
10.6. Определите стандартную энтальпию реакции взаимодействия метана с оксидом углерода (IV), если стандартные энтальпии образования метана, оксида углерода (IV) и оксида углерода (II) при 298 К равны соответственно: -75; -393 и -111 кДж/моль.
7. Определите стандартную энтальпию образования С2Н5ОН(ж), если стандартные энтальпии сгорания углерода, водорода и этанола при 298 К равны соответственно: -393; -286 и -1366 кДж/моль.
8. Вычислите энтальпию химической реакции 4KClO3(к)=KCl(к)+3KClO4(к) по следующим данным:
2KClO3(к)=2KCl(к)+3O2(г); ΔНо298 = — 95 кДж;
4KClO4(к)=KCl(к)+2O2(г); ΔНо298 = 9 кДж/моль.
9. Вычислите среднюю энтальпию связи P-Cl в молекуле PCl5, используя следующие термохимические уравнения:
P(к, бел.)+5/2Cl2(г)=PCl5(г); ΔНо1 = — 374,8 кДж;
P(к, бел.)=P(г); ΔНо2 = — 17,4 кДж;
Cl2(г)=2Cl(г); ΔНо3 = 242,6 кДж.
10. Вычислите среднюю энтальпию связи N-H в молекуле NH3, используя следующие термохимические уравнения:
1/2N2(г)+3/2H2(г)=NH3(г); ΔНо1 = — 46,2 кДж;
N2(г)=2N(г); ΔНо2 = 945,4 кДж;
H2(г)=2H(г); ΔНо3 = 436,0 кДж.
| Энтальпия | |
|---|---|
 |
|
| Размерность |
 |
| Единицы измерения | |
| СИ | Дж |
| СГС | эрг |
| Примечания | |
| Внесистемные единицы: калория, британская тепловая единица |
Энтальпи́я (от др.-греч. ενθαλπω — «нагреваю», также теплова́я фу́нкция[1][2], теплова́я фу́нкция Гиббса[3], теплосодержа́ние[1][3] и изобарно-изоэнтропийный потенциал[4]) — функция состояния  термодинамической системы, определяемая как сумма внутренней энергии
термодинамической системы, определяемая как сумма внутренней энергии  и произведения давления
и произведения давления  на объём
на объём  [1][5][6][K 1]:
[1][5][6][K 1]:
- (Определение энтальпии)

Из уравнения для дифференциала внутренней энергии[9][10]:
- (Дифференциал внутренней энергии)

где  — термодинамическая температура, а
— термодинамическая температура, а  — энтропия, следует выражение для дифференциала энтальпии[3][11][K 2]:
— энтропия, следует выражение для дифференциала энтальпии[3][11][K 2]:
- (Дифференциал энтальпии)

которое является полным дифференциалом функции  [K 3]. Она представляет собой термодинамический потенциал[⇨] относительно естественных независимых переменных — энтропии, давления и, возможно, числа частиц[⇨] и других переменных состояния [⇨].
[K 3]. Она представляет собой термодинамический потенциал[⇨] относительно естественных независимых переменных — энтропии, давления и, возможно, числа частиц[⇨] и других переменных состояния [⇨].
Понятие энтальпии существенно дополняет математический аппарат термодинамики[⇨] и гидродинамики[⇨].
Важно, что в изобарном процессе при постоянном изменение энтальпии

равное сумме изменения внутренней энергии  и совершённой системой работы
и совершённой системой работы  , в силу первого начала термодинамики равно количеству теплоты
, в силу первого начала термодинамики равно количеству теплоты  , сообщенной системе. Это свойство энтальпии позволяет использовать её для вычисления тепловыделения при различных изобарных процессах, например, химических[⇨].
, сообщенной системе. Это свойство энтальпии позволяет использовать её для вычисления тепловыделения при различных изобарных процессах, например, химических[⇨].
Отношение малого количества теплоты,  переданного системе в изобарном процессе, к изменению температуры
переданного системе в изобарном процессе, к изменению температуры  является теплоёмкостью при постоянном давлении[K 4][20]:
является теплоёмкостью при постоянном давлении[K 4][20]:

Это экспериментально измеримая величина, и из её измерений находят температурную зависимость энтальпии[⇨].
Энтальпия — экстенсивная величина: для составной системы она равна сумме энтальпий её независимых частей. Как и внутренняя энергия, энтальпия определяется с точностью до произвольного постоянного слагаемого.


Первое упоминание об использовании термина «энтальпия» в его современном значении[21]
История вопроса[править | править код]
Понятие энтальпии было введено и развито Дж. В. Гиббсом[22][23][24] в 1875 году в классической работе «О равновесии гетерогенных веществ». Для обозначения этого понятия Гиббс использовал термин «тепловая функция при постоянном давлении»[25][26].
Автором термина «энтальпия» в его современном значении считают Х. Камерлинг-Оннеса. Впервые о его авторстве упоминает работа 1909 года[27][28] в связи с обсуждением сохранения энтальпии в эффекте Джоуля — Томсона[⇨], хотя в печатных публикациях самого Камерлинг-Оннеса это слово не встречается[29]. Что же касается буквенного обозначения , до 1920-х годов оно использовалось для количества теплоты вообще. Определение физической величины строго как энтальпии или «теплосодержания при постоянном давлении» было официально предложено Альфредом У. Портером в 1922 году[23].
Энтальпия как термодинамический потенциал[править | править код]
Поскольку внутренняя энергия является термодинамическим потенциалом относительно энтропии и объёма[30],
определение энтальпии можно рассматривать как преобразование Лежандра для перехода от потенциала относительно переменных  к таковому относительно переменных
к таковому относительно переменных  Как и для любого термодинамического потенциала, естественные независимые переменные
Как и для любого термодинамического потенциала, естественные независимые переменные  совокупно с производными энтальпии по этим переменным позволяют выразить любой термодинамический параметр системы, поэтому задание термодинамического потенциала является самым общим способом задания уравнения состояния[31].
совокупно с производными энтальпии по этим переменным позволяют выразить любой термодинамический параметр системы, поэтому задание термодинамического потенциала является самым общим способом задания уравнения состояния[31].
Из выражения для дифференциала энтальпии получаются ещё два уравнения состояния, непосредственно выражающие температуру и объём через энтальпию и давление[32]:

Если известна энтальпия, другие термодинамические потенциалы — внутренняя энергия , свободная энергия Гельмгольца  и энергия Гиббса
и энергия Гиббса  — могут быть получены с помощью преобразования Лежандра:
— могут быть получены с помощью преобразования Лежандра:


Из равных друг другу смешанных производных энтальпии выводятся две термодинамические производные, связанные третьим соотношением Максвелла[33]:

Через вторые производные энтальпии выражаются ещё две термодинамические производные:

Первая из этих производных характеризует теплоёмкость при постоянном давлении  вторая — адиабатическую сжимаемость.
вторая — адиабатическую сжимаемость.
Метод якобианов позволяет получить тождества, аналогичные соотношениям Бриджмена, для выражения любых термодинамических производных через приведённые производные энтальпии.
Зависимость энтальпии от числа частиц[править | править код]
Для состоящей из одинаковых частиц открытой системы число частиц  может быть переменным[K 5]. В этом случае выражения для дифференциалов внутренней энергии и энтальпии обобщаются следующим образом[35][36]:
может быть переменным[K 5]. В этом случае выражения для дифференциалов внутренней энергии и энтальпии обобщаются следующим образом[35][36]:

где  — химический потенциал, который равен энергии Гиббса
— химический потенциал, который равен энергии Гиббса  [37], приходящейся на одну частицу[38]:
[37], приходящейся на одну частицу[38]:  . Если частицы не рождаются и не уничтожаются в рассматриваемом процессе, можно характеризовать их количество, например, (переменной) массой тела
. Если частицы не рождаются и не уничтожаются в рассматриваемом процессе, можно характеризовать их количество, например, (переменной) массой тела  и химический потенциал также относить к единице массы. В этом случае вклад от изменения массы вещества в дифференциалы энергии и энтальпии описывается членом
и химический потенциал также относить к единице массы. В этом случае вклад от изменения массы вещества в дифференциалы энергии и энтальпии описывается членом  , где модифицированный химический потенциал равен удельной (отнесенной к единице массы) энергии Гиббса:
, где модифицированный химический потенциал равен удельной (отнесенной к единице массы) энергии Гиббса:  .
.

Схема контрольного объёма. За малый интервал времени  изменение массы равно
изменение массы равно 
В англоязычной литературе, особенно технической, понятие открытой системы обычно отождествляют с понятием «контрольного объёма» (англ. control volume)[39], который ограничен воображаемой неподвижной контрольной[40] поверхностью, проницаемой для вещества, но оставляющей неизменной заключённый в ней объём. В то же время закрытую систему называют «контрольной массой» (англ. control mass). Последнее название подчеркивает постоянство массы ( ), вследствие которого справедливо приведённое выше соотношение для дифференциала внутренней энергии и термодинамическое состояние системы характеризуется только двумя параметрами, например, и . С другой стороны, при постоянстве контрольного объёма (
), вследствие которого справедливо приведённое выше соотношение для дифференциала внутренней энергии и термодинамическое состояние системы характеризуется только двумя параметрами, например, и . С другой стороны, при постоянстве контрольного объёма ( ) заключённая в нём внутренняя энергия тоже характеризуется только двумя параметрами, например, энтропией и переменной массой , причём в практически важное выражение для дифференциала внутренней энергии контрольного объёма входит (удельная) энтальпия[41]:
) заключённая в нём внутренняя энергия тоже характеризуется только двумя параметрами, например, энтропией и переменной массой , причём в практически важное выражение для дифференциала внутренней энергии контрольного объёма входит (удельная) энтальпия[41]:
- Энергия контрольного объёма)

Если в системе присутствуют несколько различных веществ характеризующихся массами  и химическими потенциалами
и химическими потенциалами  , выражение для дифференциала энтальпии обобщается следующим образом[42][43]:
, выражение для дифференциала энтальпии обобщается следующим образом[42][43]:

Удельная энтальпия[править | править код]
| Удельная энтальпия | |
|---|---|
 |
|
| Размерность |
 |
| Единицы измерения | |
| СИ | Дж/кг |
| СГС | эрг/г |
| Примечания | |
| Внесистемные единицы: кал/г, кал/кг |
| Молярная (мольная) энтальпия | |
|---|---|
 |
|
| Размерность |
|
| Единицы измерения | |
| СИ |
Дж/моль ( кг/моль) кг/моль) |
| СГС |
эрг/моль ( 1 г/моль) 1 г/моль) |
| Примечания | |
| Внесистемная единица: кал/моль |
| Плотность энтальпии | |
|---|---|
 |
|
| Размерность |
 |
| Единицы измерения | |
| СИ | Дж/м3 |
| СГС | эрг/см3 |
Вместо экстенсивной величины энтальпии часто используют её отношение  её величины к массе тела , называемое удельной энтальпией. Продолжая обозначать экстенсивные величины заглавными буквами, соответствующие им удельные величины будем обозначать строчными, за исключением удельного объёма, вместо которого введём обратную к этой величине плотность:
её величины к массе тела , называемое удельной энтальпией. Продолжая обозначать экстенсивные величины заглавными буквами, соответствующие им удельные величины будем обозначать строчными, за исключением удельного объёма, вместо которого введём обратную к этой величине плотность:

Соотношение для полного дифференциала удельной энтальпии можно получить, разделив уравнение для дифференциала энтальпии на :
- (Дифференциал удельной энтальпии)

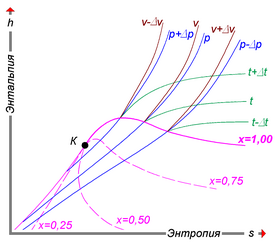
Удельную энтальпию можно представлять графически в виде  -диаграммы Молье. На диаграмме кривые (изобары) для различных значений давления задают функцию
-диаграммы Молье. На диаграмме кривые (изобары) для различных значений давления задают функцию  [44]. Большой практический интерес представляет диаграмма Молье для воды/водяного пара[45], схематически изображённая на рисунке: синие линии — изобары, зелёные — изотермы. Область ниже красной кривой соответствует двухфазной среде пара и воды. В этой области красные линии соответствуют различным значениям величины
[44]. Большой практический интерес представляет диаграмма Молье для воды/водяного пара[45], схематически изображённая на рисунке: синие линии — изобары, зелёные — изотермы. Область ниже красной кривой соответствует двухфазной среде пара и воды. В этой области красные линии соответствуют различным значениям величины  — массовой доли водяного пара — и пересекаются в критической точке K, а изобары совпадают с изотермами и являются прямыми линиями.
— массовой доли водяного пара — и пересекаются в критической точке K, а изобары совпадают с изотермами и являются прямыми линиями.
Вводят также молярную (мольную) энтальпию  , отнесённую не к массе, а к количеству вещества в теле в молях n, что удобно для приложений к химии. Молярные величины обозначают нижним индексом m. Альтернативное определение через удельную энтальпию:
, отнесённую не к массе, а к количеству вещества в теле в молях n, что удобно для приложений к химии. Молярные величины обозначают нижним индексом m. Альтернативное определение через удельную энтальпию:  , где
, где  — относительная молекулярная масса, а кг/моль =1 г/моль — коэффициент для перевода относительной молекулярной массы в молярную[46].
— относительная молекулярная масса, а кг/моль =1 г/моль — коэффициент для перевода относительной молекулярной массы в молярную[46].
Плотности внутренней энергии и энтальпии (на единицу объёма) вводят как отношение этих величин к объёму. Отдельные обозначения для этих величин здесь не вводятся, их можно выразить через удельные величины и массовую плотность:

Деление уравнения для дифференциала энергии контрольного объёма на величину контрольного объёма даёт соотношение[47]:
- (Дифференциал плотности энергии)

Плотность энергии и энтальпии идеального газа[править | править код]
Для идеального газа с постоянной теплоёмкостью плотность внутренней энергии и энтальпии простым образом выражается через давление[48]:

где  — показатель адиабаты, равный
— показатель адиабаты, равный  для одноатомного газа,
для одноатомного газа,  для фотонного газа (излучения чёрного тела)[49].
для фотонного газа (излучения чёрного тела)[49].
Энтальпия сложных термодинамических систем[править | править код]
Для термодинамических систем сложного типа, в которых термодинамическая работа[50] не сводится к работе внешних сил давления  , первое начало термодинамики, а значит и выражение для дифференциала внутренней энергии, включают вклад от термодинамической работы в виде[35][51]:
, первое начало термодинамики, а значит и выражение для дифференциала внутренней энергии, включают вклад от термодинамической работы в виде[35][51]:

где  —
—  -я обобщённая сила и
-я обобщённая сила и  — сопряжённая с ней -я обобщённая координата, во втором равенстве из общего перечня переменных выделена обобщённая сила
— сопряжённая с ней -я обобщённая координата, во втором равенстве из общего перечня переменных выделена обобщённая сила  и обобщённая координата
и обобщённая координата  . Для этого случая определение обобщённой энтальпии
. Для этого случая определение обобщённой энтальпии  [52] даёт[53]:
[52] даёт[53]:

Обобщенная энтальпии сохраняет смысл эквивалента теплоты для изобарного процесса[54][55], если не только давление, но и все остальные обобщённые силы поддерживаются постоянными:  .
.
Энтальпия образования[править | править код]
Для приложений к химии в общем случае открытых систем для полного дифференциала энтальпии получаем:

Выражение для  заимствуем из дифференциальной версии фундаментального уравнения Гиббса для внутренней энергии открытой термодинамической системы[56][57]:
заимствуем из дифференциальной версии фундаментального уравнения Гиббса для внутренней энергии открытой термодинамической системы[56][57]:

где — масса  -го независимого компонента[K 6], — химический потенциал этого компонента. Запишем уравнение Гиббса в следующем виде:
-го независимого компонента[K 6], — химический потенциал этого компонента. Запишем уравнение Гиббса в следующем виде:

Подставив это выражение в соотношение (***), получаем дифференциальную версию фундаментального уравнения Гиббса для энтальпии:
Все химические реакции сопровождаются выделением (экзотермические) или поглощением (эндотермические) тепла. Одно из приложений энтальпии основано на том, что множество химических процессов в реальных или лабораторных условиях реализуются именно при постоянном (атмосферном) давлении. Поэтому мерой теплового эффекта реакции служит изменение энтальпии ΔН в ходе химической реакции, в результате которой исходные вещества исчезают и образуются продукты реакции. В случае экзотермических реакций система теряет тепло и ΔН — величина отрицательная. В случае эндотермических реакций система поглощает тепло и ΔН — величина положительная. В частности, энтальпия образования — это количество теплоты, которое поглощается (если энтальпия образования положительна) или выделяется (если энтальпия образования отрицательна) при образовании сложного вещества из простых веществ.
Значение энтальпии образования и другие термодинамические свойства веществ приведены в справочниках[58][59].
Зависимость энтальпии от температуры[править | править код]
Во многих приложениях (но только не в качестве термодинамического потенциала!) энтальпию системы удобно представлять в виде функции  от давления и температуры . Чтобы получить выражение для дифференциала энтальпии в переменных
от давления и температуры . Чтобы получить выражение для дифференциала энтальпии в переменных  дифференциал энтропии выражается через
дифференциал энтропии выражается через  :
:

Температурная производная энтропии выражается через (измеримую) теплоёмкость при постоянном давлении  . Производная энтропии по давлению выражаются с помощью четвёртого соотношения Максвелла (G2)
. Производная энтропии по давлению выражаются с помощью четвёртого соотношения Максвелла (G2)  что даёт
что даёт  и:
и:
![{displaystyle quad mathrm {d} H=C_{P}mathrm {d} T+left[V-Tleft({frac {partial V}{partial T}}right)_{P}right]mathrm {d} P.}](https://wikimedia.org/api/rest_v1/media/math/render/svg/db8b72626dbc316bfcff5dcbb2aae49c93912304)
Для идеального газа в силу закона Гей-Люссака так что выражение в квадратных скобках равно нулю, и энтальпия идеального газа зависит только от температуры. Если к тому же идеальный газ имеет постоянную темплоёмкость, его энтальпия линейно зависит от температуры[60]:
так что выражение в квадратных скобках равно нулю, и энтальпия идеального газа зависит только от температуры. Если к тому же идеальный газ имеет постоянную темплоёмкость, его энтальпия линейно зависит от температуры[60]:
- (Энтальпия идеального газа)

где  — внутренняя энергия молекулы при нулевой температуре[K 7],
— внутренняя энергия молекулы при нулевой температуре[K 7],  — масса молекулы. Удельная энтальпии выражена через удельную теплоёмкость
— масса молекулы. Удельная энтальпии выражена через удельную теплоёмкость  отнесённую к единице массы.
отнесённую к единице массы.
Для реальных систем изменение энтальпии при изменении температуры в изобарическом процессе практически удобно рассчитывать, если известна теплоёмкость при постоянном давлении  (например, в виде ряда
(например, в виде ряда  по степеням с эмпирическими коэффициентами[61][62]):
по степеням с эмпирическими коэффициентами[61][62]):

Поскольку разности энтальпий продуктов химической реакции и исходных веществ определяет тепловой эффект химической реакции[⇨], разность теплоёмкостей продуктов реакции и исходных веществ определяет зависимость теплового эффекта реакции от температуры (термохимический закон Кирхгофа).
Сохранение энтальпии в эффекте Джоуля — Томсона[править | править код]
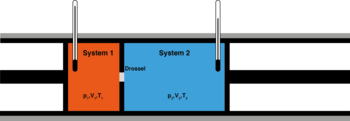
Рис. 2 — Сохранение энтальпии в эффекте Джоуля — Томсона. Изменение энергии газа в ходе этого процесса равно работе:  . Из определения энтальпии (
. Из определения энтальпии ( ) следует, что
) следует, что 
Сохранение энтальпии в процессе Джоуля — Томсона привлекается для количественного описания эффекта. Схема процесса представлена на рисунке 2. Левый поршень, вытесняя газ под давлением  из объёма
из объёма  , совершает над ним работу
, совершает над ним работу  . Пройдя через дроссель и расширяясь в объём
. Пройдя через дроссель и расширяясь в объём  , газ совершает работу
, газ совершает работу  над правым поршнем. Суммарная работа
над правым поршнем. Суммарная работа  , совершенная над газом, равна изменению его внутренней энергии
, совершенная над газом, равна изменению его внутренней энергии  , так что энтальпия сохраняется: [63][64]
, так что энтальпия сохраняется: [63][64]
Из уравнения для дифференциала энтальпии выводится выражение для коэффициента Джоуля — Томсона  , который связывает малые изменения температуры и давления в этом процессе. Приравнивание нулю дифференциала (сохраняющейся) энтальпии в переменных
, который связывает малые изменения температуры и давления в этом процессе. Приравнивание нулю дифференциала (сохраняющейся) энтальпии в переменных  даёт[65][66]
даёт[65][66] ![{displaystyle C_{P}mathrm {d} T+left[V-Tleft({frac {partial V}{partial T}}right)_{P}right]mathrm {d} P=0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/10e30be2110fa951c716fb887b72bb50a0fe82b4) и
и

а выражение для дифференциала энтальпии в переменных  даёт связь между изменениями давления и энтропии:
даёт связь между изменениями давления и энтропии:

В процессе Джоуля — Томсона давление всегда убывает, следовательно, энтропия возрастает.
Полная энергия и полная энтальпия[править | править код]
| Полная энтальпия (удельная) (энтальпия торможения) |
|
|---|---|
 |
|
| Размерность |
|
| Единицы измерения | |
| СИ | Дж/кг |
| СГС | эрг/г |
| Примечания | |
| Зависит от выбора системы отсчёта |
Для движущихся тел помимо внутренней энергии, включающей кинетическую энергию теплового движения составляющих тело частиц (измеренную в системе координат, в которой тело как целое покоится), вводят также его полную энергию в системе координат, относительно которой тело движется со скоростью  . Обычно полная энергия тела есть просто сумма его внутренней и кинетической энергий
. Обычно полная энергия тела есть просто сумма его внутренней и кинетической энергий  Более общий и строгий подход[⇨] определяет не полную энергию, а её дифференциал[67]:
Более общий и строгий подход[⇨] определяет не полную энергию, а её дифференциал[67]:
- (Дифференциал полной энергии)

где  – импульс тела и точка между векторами означает их скалярное произведение. В полную энтальпию
– импульс тела и точка между векторами означает их скалярное произведение. В полную энтальпию  также включается кинетическая энергия. Имеющие большое значение для физики сплошных сред удельная полная энергия
также включается кинетическая энергия. Имеющие большое значение для физики сплошных сред удельная полная энергия  и удельная полная энтальпия
и удельная полная энтальпия  (обычно называемая просто «полная энтальпия» или, особенно в технических науках, «энтальпия торможения»[⇨]) даются формулами:
(обычно называемая просто «полная энтальпия» или, особенно в технических науках, «энтальпия торможения»[⇨]) даются формулами:

Обобщение дифференциала плотности энергии для полной энергии принимает вид[47]:
- (Дифференциал плотности полной энергии)

Релятивистская энтальпия[править | править код]
| Полная энтальпия (инвариантная релятивистская) |
|
|---|---|
 |
|
| Размерность |
|
| Единицы измерения | |
| СИ | Дж |
| СГС | эрг |
| Примечания | |
| Лоренц-инвариант |
| Полная энтальпия (релятивистская) |
|
|---|---|
 |
|
| Размерность |
|
| Единицы измерения | |
| СИ | Дж |
| СГС | эрг |
| Примечания | |
Образует 4-вектор вместе с импульсом  |
Если скорость тела сравнима по величине со скоростью света  , термодинамика строится с учётом специальной теории относительности)[67]. При этом используется инвариантная энтальпия , которая представляет собой полную энтальпию, определённую в движущейся вместе с телом системе отсчета; все величины в этой системе отсчёта обозначаем нижним индексом «0».
, термодинамика строится с учётом специальной теории относительности)[67]. При этом используется инвариантная энтальпия , которая представляет собой полную энтальпию, определённую в движущейся вместе с телом системе отсчета; все величины в этой системе отсчёта обозначаем нижним индексом «0».
Релятивистская полная энергия  , включает энергию покоя всех частиц тела и учитывает релятивистскую зависимость их энергии
, включает энергию покоя всех частиц тела и учитывает релятивистскую зависимость их энергии  от импульса
от импульса  , а именно: 1) энергия и импульс образуют 4-вектор, 2) величина
, а именно: 1) энергия и импульс образуют 4-вектор, 2) величина  является Лоренц-инвариантом и 3) величина
является Лоренц-инвариантом и 3) величина  являются скоростью частицы.
являются скоростью частицы.
В неподвижной системе отсчета энтальпия и импульс движущегося тела[68][67]

образуют 4-вектор, а инвариантная энтальпии в движущейся с телом системы отсчёта даётся инвариантной функцией этого 4-вектора:

Именно полная энтальпия (а не энергия) релятивистского тела оказывается аналогом энергии релятивистской частицы. Давление  лоренц-инвариантно, а преобразование объёма :
лоренц-инвариантно, а преобразование объёма :

является следствием Лоренцева сокращения. Уравнение релятивистской термодинамики даётся выражением[68]:

Оно позволяет решить любой вопрос термодинамики движущихся систем, если известна функция  [68]. В частности, из выражения для Дифференциала полной энергии
[68]. В частности, из выражения для Дифференциала полной энергии  можно получить выражение для малого количества теплоты[67]:
можно получить выражение для малого количества теплоты[67]: 
Энтальпия в гидродинамике[править | править код]
| Механика сплошных сред |
|---|
 |
| Сплошная среда |
|
Классическая механика Закон сохранения массы · Закон сохранения импульса |
|
Теория упругости Напряжение · Тензор · Твёрдые тела · Упругость · Пластичность · Закон Гука · Реология · Вязкоупругость |
|
Гидродинамика Жидкость · Гидростатика · Гидродинамика · Вязкость · Ньютоновская жидкость · Неньютоновская жидкость · Поверхностное натяжение |
|
Основные уравнения Уравнение непрерывности · Уравнение Эйлера · Уравнение Громеки — Лэмба · Уравнение Бернулли · Уравнения Навье — Стокса · Уравнение вихря · Уравнение диффузии · Закон Гука |
| См. также: Портал:Физика |
Энтальпия играет большую роль в гидродинамике, науке о движениях жидкостей и газов (в гидродинамике газы тоже называют жидкостями). Течения идеальной жидкости (то есть без вязкости и теплопроводности) описываются следующими уравнениями в частных производных[69]:
- (Уравнение непрерывности)
- (Уравнение Эйлера)


где  — плотность;
— плотность;  — скорость;
— скорость;  — давление;
— давление;  — время;
— время;  — векторный оператор частного дифференцирования по координатам
— векторный оператор частного дифференцирования по координатам  ; точка между векторами в круглых скобках означает их скалярное произведение, а
; точка между векторами в круглых скобках означает их скалярное произведение, а  — ускорение силы тяжести, выраженное через гравитационный потенциал
— ускорение силы тяжести, выраженное через гравитационный потенциал  Уравнение для Дифференциала удельной энтальпии даёт:
Уравнение для Дифференциала удельной энтальпии даёт:  что позволяет выразить уравнение Эйлера через энтальпию:
что позволяет выразить уравнение Эйлера через энтальпию:
- (Уравнение Эйлера, выраженное через энтальпию)

Такое представление обладает значительными преимуществами, поскольку в силу «адабатичности» течения идеальной жидкости, задаваемого уравнением сохранения энтропии:

член в уравнении Эйлера, связанный с градиентом энтропии, во многих случаях не даёт вклада в рассчитываемые эффекты.
Поток энергии[править | править код]
Выражение для дифференциала плотности полной энергии позволяет получить скорость изменения последней[47]:

Интеграл Бернулли[править | править код]
Из приведённых здесь термодинамических соотношений для энтальпии следует простой вывод интеграла Бернулли и в наиболее общей его форме. Закон утверждает, что вдоль линии тока для стационарного течения идеальной жидкости[70] сохраняется следующая величина:

где  — гравитационный потенциал (равный
— гравитационный потенциал (равный  для однородной силы тяжести,
для однородной силы тяжести,  — ускорение свободного падения,
— ускорение свободного падения,  — вертикальная координата).
— вертикальная координата).
Вывод закона Бернулли из уравнения Эйлера и термодинамических соотношений
1. В уравнении Эйлера для стационарного ( ) движения идеальной жидкости в поле силы тяжести[69] ускорение силы тяжести можно выразить через гравитационный потенциал
) движения идеальной жидкости в поле силы тяжести[69] ускорение силы тяжести можно выразить через гравитационный потенциал  (для однородного поля
(для однородного поля  ).
).
2. Скалярное произведение этого уравнения на единичный вектор  касательный к линии тока даёт:
касательный к линии тока даёт:

так как произведение градиента на единичный вектор даёт производную по направлению 
3. Выражение для Дифференциала удельной энтальпии даёт:
- так что


В стационарном течении идеальной жидкости все частицы, движущиеся вдоль данной линии тока, имеют одинаковую энтропию[71] ( ), поэтому вдоль линии тока:
), поэтому вдоль линии тока:

См. также[править | править код]
- Тепловой эффект химической реакции
- Удельная теплота плавления
Комментарии[править | править код]
- ↑ В России определение энтальпии как суммы закреплено действующими стандартами[7][8].
- ↑ Это соотношение носит название дифференциальной формы фундаментального уравнения Гиббса для энтальпии закрытой термодеформационной системы[12][13][14].
- ↑ Энтальпию, заданную в виде функции её естественных независимых переменных, называют интегральной формой фундаментального уравнения Гиббса[15][16][17] для энтальпии закрытой термодеформационной системы[12][18][19].
- ↑ В термодинамике при написании частных производных внизу справа указывают переменные, который при вычислении производной считают постоянным. Причина в том, что в термодинамике для одной и той же функции используют различные наборы независимых переменных, которые, во избежание неопределённости, приходится перечислять.
- ↑ Число частиц в закрытой системе тоже может быть переменным, например числе фотонов равновесного излучения в полости с абсолютно чёрными стенками[34].
- ↑ Использование масс независимых компонентов, а не масс составляющих систему веществ, позволяет учитывать химические превращения в системе без явного рассмотрения протекающих в ней химических реакций (см. статью Химическая термодинамика).
- ↑ Энергия включает в себя энергию химической связи и вносит значительный вклад в энтальпию образования[⇨] газообразных сложных веществ

Примечания[править | править код]
- ↑ 1 2 3 Энтальпия // Большая российская энциклопедия. Том 35. Москва, 2017, стр. 396.
- ↑ Ландау Л. Д., Лифшиц Е. М. Статистическая физика. Часть 1, 2002, §14. Тепловая функция.
- ↑ 1 2 3 Зубарев Д. Н., Энтальпия, 1992.
- ↑ Горшков В. И., Кузнецов И. А., Основы физической химии, 2009, с. 111.
- ↑ Enthalpy, H // IUPAC Gold Book.
- ↑ Термодинамика. Основные понятия. Терминология. Буквенные обозначения величин, 1984, с. 13.
- ↑ §113-04-21. Энтальпия (Н)//ГОСТ IEC 60050-113-2015 (2015). Дата обращения: 1 декабря 2018.
- ↑ §54. Энтальпия(теплосодержание)//ГОСТ Р 57700.4-2017 (2017). Дата обращения: 1 декабря 2018.
- ↑ Зубарев Д. Н., Термодинамика, 1992.
- ↑ Ландау Л. Д., Лифшиц Е. М. Статистическая физика. Часть 1, 2002, Уравнение (12.3).
- ↑ Ландау Л. Д., Лифшиц Е. М. Статистическая физика. Часть 1, 2002, Уравнение (16.3).
- ↑ 1 2 Белов Г. В., Термодинамика, ч. 1, 2017, с. 155.
- ↑ Степановских Е. И. и др., Химическая термодинамика в вопросах и ответах, 2014, с. 37.
- ↑ Мечковский Л. А., Блохин А. В., Химическая термодинамика, ч. 1, 2012, с. 124.
- ↑ Борщевский А. Я., Физическая химия, т. 1, 2017, с. 312.
- ↑ Воронин Г. Ф., Основы термодинамики, 1987, с. 76.
- ↑ Мюнстер А., Химическая термодинамика, 2002, с. 90—91.
- ↑ Белов Г. В., Термодинамика, ч. 2, 2016, с. 23.
- ↑ Зарубин Д. П., Физическая химия, 2017, с. 45.
- ↑ Сивухин Д. В., Термодинамика и молекулярная физика, 2005, с. 67.
- ↑ Dalton, 1909.
- ↑ Гиббс Дж. В., Термодинамические работы, 1950, Примечание 3, с. 448.
- ↑ 1 2 Howard, 2002, с. 697.
- ↑ Ахметов Б. В. и др. Физическая и коллоидная химия, 1986, с. 64.
- ↑ Гиббс Дж. В., Термодинамика. Статистическая механика, 1982, с. 96, 510.
- ↑ Henderson, Douglas; Eyring, Henry; Jost, Wilhelm. Physical Chemistry: An Advanced Treatise (неопр.). — Academic Press, 1967. — С. 29.
- ↑ Dalton, 1909, с. 863.
- ↑ Laidler (англ.) (рус.; Keith. The World of Physical Chemistry (англ.). — Oxford University Press, 1995. — P. 110.
- ↑ Van Ness, 2003, с. 486.
- ↑ Зубарев Д. Н., Потенциал термодинамический, 1994, с. 89.
- ↑ Сивухин Д. В., Термодинамика и молекулярная физика, 2005, §45. Термодинамические функции.
- ↑ Ландау Л. Д., Лифшиц Е. М. Статистическая физика. Часть 1, 2002, Уравнение (14.4).
- ↑ Беляев Н. М., Термодинамика, 1987, с. 126.
- ↑ Ландау Л. Д., Лифшиц Е. М. Статистическая физика. Часть 1, 2002, §63. Чёрное излучение.
- ↑ 1 2 Зубарев Д. Н., Термодинамика, 1992.
- ↑ Ландау Л. Д., Лифшиц Е. М. Статистическая физика. Часть 1, 2002, Уравнения (24.5–7).
- ↑ Ландау Л. Д., Лифшиц Е. М. Статистическая физика. Часть 1, 2002, Уравнение (15.7).
- ↑ Ландау Л. Д., Лифшиц Е. М. Статистическая физика. Часть 1, 2002, Уравнение (24.11).
- ↑ Automotive Encyclopedia, 2015, §1.1.2.2. Open dynamic system, с. 27.
- ↑ Белов Г. В., Термодинамика, ч. 1, 2017, с. 11.
- ↑ Automotive Encyclopedia, 2015, Уравнение (15), с. 28.
- ↑ Мюнстер А., Химическая термодинамика, 2002, с. 103.
- ↑ Кубо Р., Термодинамика, 1970, с. 24—25.
- ↑ Поль Р. В., Механика, акустика и учение о теплоте, 2013, с. 446.
- ↑ Поль Р. В., Механика, акустика и учение о теплоте, 2013, с. 449–451.
- ↑ Quantities, Units and Symbols in Physical Chemistry (англ.). IUPAC (2015). Дата обращения: 7 декабря 2018. Архивировано из оригинала 11 февраля 2014 года.
- ↑ 1 2 3 Ландау Л. Д., Лифшиц Е. М. Гидродинамика, 2015, §6. Поток энергии.
- ↑ Ландау Л. Д., Лифшиц Е. М. Статистическая физика. Часть 1, 2002, Уравнения (42.5), (43.2) и (43.4).
- ↑ Ландау Л. Д., Лифшиц Е. М. Статистическая физика. Часть 1, 2002, §63.
- ↑ Зубарев Д. Н., Работа в термодинамике, 1994.
- ↑ Сивухин Д. В., Термодинамика и молекулярная физика, 2005, §12.
- ↑ Сивухин Д. В., Термодинамика и молекулярная физика, 2005, Уравнение (45.21).
- ↑ Сивухин Д. В., Термодинамика и молекулярная физика, 2005, Уравнение (45.25).
- ↑ Борщевский А. Я., Физическая химия, т. 1, 2017, с. 121.
- ↑ Базаров И. П., Термодинамика, 2010, с. 113.
- ↑ Артемов А. В., Физическая химия, 2013, с. 23.
- ↑ Ипполитов Е. Г. и др., Физическая химия, 2005, с. 35.
- ↑ Search for Species Data by Chemical Formula (англ.). Дата обращения: 3 декабря 2018.
- ↑ Термодинамические свойства. Дата обращения: 3 декабря 2018.
- ↑ Ландау Л. Д., Лифшиц Е. М. Статистическая физика. Часть 1, 2002, Уравнение (43.4).
- ↑ Пригожин И., Дефэй Р., Химическая термодинамика, 2009, с. 51.
- ↑ Алабовский А. Н., Недужий И. А., Техническая термодинамика и теплопередача, 1990, с. 25—26.
- ↑ Сивухин Д. В., Термодинамика и молекулярная физика, 2005, Уравнение (19.3).
- ↑ Ландау Л. Д., Лифшиц Е. М. Статистическая физика. Часть 1, 2002, Уравнение (18.1).
- ↑ Сивухин Д. В., Термодинамика и молекулярная физика, 2005, Уравнение (46.1).
- ↑ Ландау Л. Д., Лифшиц Е. М. Статистическая физика. Часть 1, 2002, Уравнение (18.2).
- ↑ 1 2 3 4 Зубарев Д. Н., Релятивистская термодинамика, 1994.
- ↑ 1 2 3 Каллен Г., Горвиц Дж., Релятивистская термодинамика, 1972.
- ↑ 1 2 Ландау Л. Д., Лифшиц Е. М. Гидродинамика, 2015, Уравнение (2.4).
- ↑ Вишневецкий С. Л., Бернулли уравнение, 1988, с. 187.
- ↑ Седов Л. И. Механика сплошной среды, 1970, Глава VII. §2. Функция давления.
Литература[править | править код]
- Алабовский А. Н., Недужий И. А. Техническая термодинамика и теплопередача. — 3-е изд., пераб. и доп. — Киев: Выща школа, 1990. — 256 с. — ISBN 5-11-001997-5.
- Ансельм А. И. Основы статистической физики и термодинамики. — 2-е изд., стереотип. — СПб.: Лань, 2007. — 427 с. — (Учебники для вузов. Специальная литература). — ISBN 978-5-8114-0756-9.
- Артемов А. В. Физическая химия. — М.: Академия, 2013. — 288 с. — (Бакалавриат). — ISBN 978-5-7695-9550-9.
- Ахметов Б. В., Новиченко Ю. П., Чапурин В. И. Физическая и коллоидная химия. — Л.: Химия, 1986. — 320 с.
- Базаров И. П. Термодинамика. — 5-е изд. — СПб.—М.—Краснодар: Лань, 2010. — 384 с. — (Учебники для вузов. Специальная литература). — ISBN 978-5-8114-1003-3.
- Белов Г. В. Термодинамика. Часть 1. — 2-е изд., испр. и доп. — М.: Юрайт, 2017. — 265 с. — (Бакалавр. Академический курс). — ISBN 978-5-534-02731-0.
- Белов Г. В. Термодинамика. Часть 2. — М.: Юрайт, 2016. — 249 с. — (Бакалавриат). — ISBN 978-5-9916-7252-8.
- Беляев Н. М. Термодинамика. — Киев: Вища школа, 1987. — 344 с.
- Болгарский А. В., Мухачев Г. А., Щукин В. К. Термодинамика и теплопередача. — 2-е изд., перераб. и доп. — М.: Высшая школа, 1975. — 496 с.
- Борщевский А. Я. Физическая химия. Том 1 online. Общая и химическая термодинамика. — М.: Инфра-М, 2017. — 868 с. — (Высшее образование: Бакалавриат). — ISBN 978-5-16-104227-4.
- Буданов В. В., Максимов А. И. Химическая термодинамика / Под ред. О. И. Койфмана. — 3-е изд., стер. — СПб.: Лань, 2017. — 320 с. — (Учебники для вузов. Специальная литература). — ISBN 978-5-8114-2271-5.
- Вишневецкий С. Л. Бернулли уравнение // Физическая энциклопедия. — Большая Российская энциклопедия, 1988. — Т. 5: Стробоскопические приборы — Яркость. — С. 187.
- Воронин Г. Ф. Основы термодинамики. — М.: Изд-во Моск. ун-та, 1987. — 192 с.
- Гамбург Ю. Д. Химическая термодинамика. — М.: Лаборатория знаний, 2016. — 237 с. — (Учебник для высшей школы). — ISBN 978-5-906828-74-3.
- Герасимов Я. И., Древинг В. П., Еремин Е. Н. и др. Курс физической химии / Под общ. ред. Я. И. Герасимова. — 2-е изд., испр. — М.: Химия, 1970. — Т. 1. — 592 с.
- Гиббс Дж. В. Термодинамические работы / Пер. с англ. под ред. проф. В. К. Семенченко. — М. — Л.: Гостехиздат, 1950. — 492 с. — (Классики естествознания).
- Гиббс Дж. В. Термодинамика. Статистическая механика / Отв. ред. Д. Н. Зубарев. — М.: Наука, 1982. — 584 с. — (Классики науки).
- Горшков В. И., Кузнецов И. А. Основы физической химии. — 3-е изд. — М.: Бином. Лаборатория знаний, 2009. — 408 с. — ISBN 978-5-94774-375-3.
- Еремин В. В., Каргов С. И., Успенская И. А. и др. Основы физической химии. Теория и задачи. — М.: Экзамен, 2005. — 481 с. — (Классический университетский учебник). — ISBN 5-472-00834-4.
- Зарубин Д. П. Физическая химия. — М.: Инфра-М, 2017. — 474 с.
- Зубарев, Д. Н. Потенциал термодинамический // Физическая энциклопедия / Гл. ред. А. М. Прохоров. — М.: Большая российская энциклопедия, 1994. — Т. 4: Пойнтинга-Робертсона эффект — Стримеры. — С. 89—91. — 704 с. — ISBN 5-85270-087-8.
- Зубарев, Д. Н. Работа в термодинамике // Физическая энциклопедия / Гл. ред. А. М. Прохоров. — М.: Большая российская энциклопедия, 1994. — Т. 4: Пойнтинга-Робертсона эффект — Стримеры. — С. 193. — 704 с. — ISBN 5-85270-087-8.
- Зубарев, Д. Н. Релятивистская термодинамика // Физическая энциклопедия / Гл. ред. А. М. Прохоров. — М.: Большая российская энциклопедия, 1994. — Т. 4: Пойнтинга-Робертсона эффект — Стримеры. — С. 333—334. — 704 с. — ISBN 5-85270-087-8.
- Зубарев Д. Н. Термодинамика // Физическая энциклопедия / Ред.А. М. Прохоров. — М.: Большая Советская Энциклопедия, 1992. — Т. 5. — С. 83—87.
- Зубарев Д. Н. Энтальпия // Физическая энциклопедия / Ред. А. М. Прохоров. — М.: Большая Советская Энциклопедия, 1992. — Т. 5. — С. 616.
- Ипполитов Е. Г., Артемов А. В., Батраков В.В. Физическая химия / Под ред. Е. Г. Ипполитова. — М.: Академия, 2005. — 448 с. — (Высшее профессиональное образование). — ISBN 978-5-7695-1456-6.
- Каллен Г., Горвиц Дж. Релятивистская термодинамика // Успехи Физических Наук : журнал. — 1972. — Т. 107, вып. 7. — С. 489—502. — doi:10.3367/UFNr.0107.197207g.0489.
- Колесников И. М., Винокуров В. А. Термодинамика физико-химических процессов. — 2-е, перераб. и доп. — М.: Нефть и газ, 2005. — 480 с. — ISBN 5-7246-0351-9.
- Кубо Р. Термодинамика. — М.: Мир, 1970. — 304 с.
- Ландау Л. Д., Лифшиц Е. М. Гидродинамика. — Издание 6-е, исправленное. — М.: Физматлит, 2015. — 728 с. — («Теоретическая физика», том VI). — ISBN 978-5-9221-1625-1.
- Ландау Л. Д., Лифшиц Е. М. Статистическая физика. Часть 1. — Издание 5-е. — М.: Физматлит, 2002. — 616 с. — («Теоретическая физика», том V). — ISBN 5-9221-0054-8.
- Мечковский Л. А., Блохин А. В. Химическая термодинамика. В двух частях. Часть 1. Феноменологическая термодинамика. Основные понятия, фазовые равновесия. — Минск: Издательство БГУ, 2012. — 141 с. — ISBN 978-985-518-635-0.
- Мюнстер А. Химическая термодинамика / Пер. с нем. под. ред. чл.-корр. АН СССР Я. И. Герасимова. — 2-е изд., стереотип. — М.: УРСС, 2002. — 296 с. — ISBN 5-354-00217-6.
- Новиков И. И. Термодинамика. — 2-е изд., испр. — СПб.: Лань, 2009. — 592 с. — (Учебники для вузов. Специальная литература). — ISBN 978-5-8114-0987-7.
- Поль Р. В. Механика, акустика и учение о теплоте. — Рипол Классик, 2013. — 490 с. — ISBN 5458431251, 9785458431255.
- Пригожин И., Дефэй Р. Химическая термодинамика / Пер. с англ. под ред. В. А. Михайлова. — 2-е изд.. — М.: Бином. Лаборатория знаний, 2009. — 533 с. — (Классика и современность. Естествознание). — ISBN 978-5-9963-0201-7.
- Савельев И. В. Курс общей физики. — М.: КноРус, 2012. — Т. 1. Механика. Молекулярная физика и термодинамика. — 528 с. — ISBN 9785406025888.
- Свиридов В. В., Свиридов А. В. Физическая химия. — СПб.: Лань, 2016. — 597 с. — ISBN 978-5-8114-2262-3.
- Седов Л. И. Механика сплошной среды. — М.: Наука, 1970. — Т. 2. — 568 с.
- Сивухин Д. В. Общий курс физики. — Издание 5-е, исправленное. — М.: Физматлит, 2005. — Т. II. Термодинамика и молекулярная физика. — 544 с. — ISBN 5-9221-0601-5.
- Степановских Е. И., Брусницына Л. А., Маскаева Л. Н. Химическая термодинамика в вопросах и ответах. — Екатеринбург: Уральский издательско-полиграфический центр, 2014. — 221 с. — ISBN 978-5-4430-0061-9.
- Сычёв В. В. Сложные термодинамические системы. — 5-е изд., перераб. и доп. — М.: Издательский дом МЭИ, 2009. — 296 с. — ISBN 978-5-383-00418-0.
- Термодинамика. Основные понятия. Терминология. Буквенные обозначения величин / Отв. ред. И. И. Новиков. — АН СССР. Комитет научно-технической терминологии. Сборник определений. Вып. 103. — М.: Наука, 1984. — 40 с.
- Хаазе Р. Термодинамика необратимых процессов / Пер. с нем. под ред. А. В. Лыкова. — М.: Мир, 1967. — 544 с.
- Хачкурузов Г. А. Основы общей и химической термодинамики. — М.: Высшая школа, 1979. — 268 с.
- Dalton J. P. Researches on the Joule-Kelvin effect, especially at low temperatures. I. Calculations for hydrogen // KNAW Proceedings. — 1909. — Т. 11. — С. 863—873.
- Howard Irmgard K. H Is for Enthalpy, Thanks to Heike Kamerlingh Onnes and Alfred W. Porter (англ.) // Journal of Chemical Education : журнал. — 2002. — Vol. 79, iss. 6. — P. 697—698. — ISSN 0021-9584. — doi:10.1021/ed079p697.
- Van Ness Hendrick C. H Is for Enthalpy (англ.) // Journal of Chemical Education : журнал. — 2003. — Vol. 80, iss. 5. — P. 486. — ISSN 0021-9584. — doi:10.1021/ed080p486.1.
- Part 1 : Engines — Fundamentals // Encyclopedia of Automitive Engineering / Editors-in-Chief : David Crolla, David E. Foster, Toshio Kobayashi, Nicolas Vaughan. — John Wiley & Sons, 2015. — Т. 1. — 607 с. — ISBN 978-0-470-97402-5.
From Wikipedia, the free encyclopedia
In chemistry and thermodynamics, the standard enthalpy of formation or standard heat of formation of a compound is the change of enthalpy during the formation of 1 mole of the substance from its constituent elements in their reference state, with all substances in their standard states. The standard pressure value p⦵ = 105 Pa (= 100 kPa = 1 bar) is recommended by IUPAC, although prior to 1982 the value 1.00 atm (101.325 kPa) was used.[1] There is no standard temperature. Its symbol is ΔfH⦵. The superscript Plimsoll on this symbol indicates that the process has occurred under standard conditions at the specified temperature (usually 25 °C or 298.15 K). Standard states are as follows:
- For a gas: the hypothetical state the gas would assume if it obeyed the ideal gas equation at a pressure of 1 bar
- For a gaseous or solid solute present in a diluted ideal solution: the hypothetical state of concentration of the solute of exactly one mole per liter (1 M) at a pressure of 1 bar extrapolated from infinite dilution
- For a pure substance or a solvent in a condensed state (a liquid or a solid): the standard state is the pure liquid or solid under a pressure of 1 bar
For elements that have multiple allotropes, the reference state usually is chosen to be the form in which the element is most stable under 1 bar of pressure. One exception is phosphorus, for which the most stable form at 1 bar is black phosphorus, but white phosphorus is chosen as the standard reference state for zero enthalpy of formation.[2]
For example, the standard enthalpy of formation of carbon dioxide would be the enthalpy of the following reaction under the above conditions:

All elements are written in their standard states, and one mole of product is formed. This is true for all enthalpies of formation.
The standard enthalpy of formation is measured in units of energy per amount of substance, usually stated in kilojoule per mole (kJ mol−1), but also in kilocalorie per mole, joule per mole or kilocalorie per gram (any combination of these units conforming to the energy per mass or amount guideline).
All elements in their reference states (oxygen gas, solid carbon in the form of graphite, etc.) have a standard enthalpy of formation of zero, as there is no change involved in their formation.
The formation reaction is a constant pressure and constant temperature process. Since the pressure of the standard formation reaction is fixed at 1 bar, the standard formation enthalpy or reaction heat is a function of temperature. For tabulation purposes, standard formation enthalpies are all given at a single temperature: 298 K, represented by the symbol ΔfH⦵
298 K.
Hess’s law[edit]
For many substances, the formation reaction may be considered as the sum of a number of simpler reactions, either real or fictitious. The enthalpy of reaction can then be analyzed by applying Hess’s Law, which states that the sum of the enthalpy changes for a number of individual reaction steps equals the enthalpy change of the overall reaction. This is true because enthalpy is a state function, whose value for an overall process depends only on the initial and final states and not on any intermediate states. Examples are given in the following sections.
Ionic compounds: Born–Haber cycle[edit]

Standard enthalpy change of formation in Born–Haber diagram for lithium fluoride. ΔHlatt corresponds to UL in the text. The downward arrow “electron affinity” shows the negative quantity –EAF, since EAF is usually defined as positive.
For ionic compounds, the standard enthalpy of formation is equivalent to the sum of several terms included in the Born–Haber cycle. For example, the formation of lithium fluoride,

may be considered as the sum of several steps, each with its own enthalpy (or energy, approximately):
- Hsub, the standard enthalpy of atomization (or sublimation) of solid lithium.
- IELi, the first ionization energy of gaseous lithium.
- B(F–F), the standard enthalpy of atomization (or bond energy) of fluorine gas.
- EAF, the electron affinity of a fluorine atom.
- UL, the lattice energy of lithium fluoride.
The sum of all these enthalpies will give the standard enthalpy of formation (ΔHf) of lithium fluoride:

In practice, the enthalpy of formation of lithium fluoride can be determined experimentally, but the lattice energy cannot be measured directly. The equation is therefore rearranged in order to evaluate the lattice energy:[3]

Organic compounds[edit]
The formation reactions for most organic compounds are hypothetical. For instance, carbon and hydrogen won’t directly react to form methane (CH4), so that the standard enthalpy of formation cannot be measured directly. However the standard enthalpy of combustion is readily measurable using bomb calorimetry. The standard enthalpy of formation is then determined using Hess’s law. The combustion of methane:

is equivalent to the sum of the hypothetical decomposition into elements followed by the combustion of the elements to form carbon dioxide (CO2) and water (H2O):



Applying Hess’s law,
![{displaystyle Delta _{text{comb}}H^{ominus }({text{CH}}_{4})=[Delta _{text{f}}H^{ominus }({text{CO}}_{2})+2Delta _{text{f}}H^{ominus }({text{H}}_{2}{text{O}})]-Delta _{text{f}}H^{ominus }({text{CH}}_{4}).}](https://wikimedia.org/api/rest_v1/media/math/render/svg/868ef759a6238c57f082b526d2599d6a04ae8c6a)
Solving for the standard of enthalpy of formation,
![{displaystyle Delta _{text{f}}H^{ominus }({text{CH}}_{4})=[Delta _{text{f}}H^{ominus }({text{CO}}_{2})+2Delta _{text{f}}H^{ominus }({text{H}}_{2}{text{O}})]-Delta _{text{comb}}H^{ominus }({text{CH}}_{4}).}](https://wikimedia.org/api/rest_v1/media/math/render/svg/eaa4ff8ecbdfc22efc596605a9e9c5b9fc178c70)
The value of  is determined to be −74.8 kJ/mol. The negative sign shows that the reaction, if it were to proceed, would be exothermic; that is, methane is enthalpically more stable than hydrogen gas and carbon.
is determined to be −74.8 kJ/mol. The negative sign shows that the reaction, if it were to proceed, would be exothermic; that is, methane is enthalpically more stable than hydrogen gas and carbon.
It is possible to predict heats of formation for simple unstrained organic compounds with the heat of formation group additivity method.
Use in calculation for other reactions[edit]
The standard enthalpy change of any reaction can be calculated from the standard enthalpies of formation of reactants and products using Hess’s law. A given reaction is considered as the decomposition of all reactants into elements in their standard states, followed by the formation of all products. The heat of reaction is then minus the sum of the standard enthalpies of formation of the reactants (each being multiplied by its respective stoichiometric coefficient, ν) plus the sum of the standard enthalpies of formation of the products (each also multiplied by its respective stoichiometric coefficient), as shown in the equation below:[4]

If the standard enthalpy of the products is less than the standard enthalpy of the reactants, the standard enthalpy of reaction is negative. This implies that the reaction is exothermic. The converse is also true; the standard enthalpy of reaction is positive for an endothermic reaction. This calculation has a tacit assumption of ideal solution between reactants and products where the enthalpy of mixing is zero.
For example, for the combustion of methane,  :
:
![{displaystyle Delta _{text{r}}H^{ominus }=[Delta _{text{f}}H^{ominus }({text{CO}}_{2})+2Delta _{text{f}}H^{ominus }({text{H}}_{2}{text{O}})]-Delta _{text{f}}H^{ominus }({text{CH}}_{4})+2Delta _{text{f}}H^{ominus }({text{O}}_{2})].}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d13e59b354921b5d0bcc0e9da48fd3e719de3e74)
However  is an element in its standard state, so that
is an element in its standard state, so that  , and the heat of reaction is simplified to
, and the heat of reaction is simplified to
![{displaystyle Delta _{text{r}}H^{ominus }=[Delta _{text{f}}H^{ominus }({text{CO}}_{2})+2Delta _{text{f}}H^{ominus }({text{H}}_{2}{text{O}})]-Delta _{text{f}}H^{ominus }({text{CH}}_{4}),}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2bb97407d417f1efde328dead9949da6b6d6d019)
which is the equation in the previous section for the enthalpy of combustion  .
.
Key concepts for enthalpy calculations[edit]
- When a reaction is reversed, the magnitude of ΔH stays the same, but the sign changes.
- When the balanced equation for a reaction is multiplied by an integer, the corresponding value of ΔH must be multiplied by that integer as well.
- The change in enthalpy for a reaction can be calculated from the enthalpies of formation of the reactants and the products
- Elements in their standard states make no contribution to the enthalpy calculations for the reaction, since the enthalpy of an element in its standard state is zero. Allotropes of an element other than the standard state generally have non-zero standard enthalpies of formation.
Examples: standard enthalpies of formation at 25 °C[edit]
Thermochemical properties of selected substances at 298.15 K and 1 atm
Inorganic substances[edit]
| Species | Phase | Chemical formula | ΔfH⦵ /(kJ/mol) |
|---|---|---|---|
| Aluminium | Solid | Al | 0 |
| Aluminium chloride | Solid | AlCl3 | −705.63 |
| Aluminium oxide | Solid | Al2O3 | −1675.5 |
| Aluminium hydroxide | Solid | Al(OH)3 | −1277 |
| Aluminium sulphate | Solid | Al2(SO4)3 | −3440 |
| Barium chloride | Solid | BaCl2 | −858.6 |
| Barium carbonate | Solid | BaCO3 | −1216 |
| Barium hydroxide | Solid | Ba(OH)2 | −944.7 |
| Barium oxide | Solid | BaO | −548.1 |
| Barium sulfate | Solid | BaSO4 | −1473.3 |
| Beryllium | Solid | Be | 0 |
| Beryllium hydroxide | Solid | Be(OH)2 | −903 |
| Beryllium oxide | Solid | BeO | −609.4 |
| Boron trichloride | Solid | BCl3 | −402.96 |
| Bromine | Liquid | Br2 | 0 |
| Bromide ion | Aqueous | Br− | −121 |
| Bromine | Gas | Br | 111.884 |
| Bromine | Gas | Br2 | 30.91 |
| Bromine trifluoride | Gas | BrF3 | −255.60 |
| Hydrogen bromide | Gas | HBr | −36.29 |
| Cadmium | Solid | Cd | 0 |
| Cadmium oxide | Solid | CdO | −258 |
| Cadmium hydroxide | Solid | Cd(OH)2 | −561 |
| Cadmium sulfide | Solid | CdS | −162 |
| Cadmium sulfate | Solid | CdSO4 | −935 |
| Caesium | Solid | Cs | 0 |
| Caesium | Gas | Cs | 76.50 |
| Caesium | Liquid | Cs | 2.09 |
| Caesium(I) ion | Gas | Cs+ | 457.964 |
| Caesium chloride | Solid | CsCl | −443.04 |
| Calcium | Solid | Ca | 0 |
| Calcium | Gas | Ca | 178.2 |
| Calcium(II) ion | Gas | Ca2+ | 1925.90 |
| Calcium(II) ion | Aqueous | Ca2+ | −542.7 |
| Calcium carbide | Solid | CaC2 | −59.8 |
| Calcium carbonate (Calcite) | Solid | CaCO3 | −1206.9 |
| Calcium chloride | Solid | CaCl2 | −795.8 |
| Calcium chloride | Aqueous | CaCl2 | −877.3 |
| Calcium phosphate | Solid | Ca3(PO4)2 | −4132 |
| Calcium fluoride | Solid | CaF2 | −1219.6 |
| Calcium hydride | Solid | CaH2 | −186.2 |
| Calcium hydroxide | Solid | Ca(OH)2 | −986.09 |
| Calcium hydroxide | Aqueous | Ca(OH)2 | −1002.82 |
| Calcium oxide | Solid | CaO | −635.09 |
| Calcium sulfate | Solid | CaSO4 | −1434.52 |
| Calcium sulfide | Solid | CaS | −482.4 |
| Wollastonite | Solid | CaSiO3 | −1630 |
| Carbon (Graphite) | Solid | C | 0 |
| Carbon (Diamond) | Solid | C | 1.9 |
| Carbon | Gas | C | 716.67 |
| Carbon dioxide | Gas | CO2 | −393.509 |
| Carbon disulfide | Liquid | CS2 | 89.41 |
| Carbon disulfide | Gas | CS2 | 116.7 |
| Carbon monoxide | Gas | CO | −110.525 |
| Carbonyl chloride (Phosgene) | Gas | COCl2 | −218.8 |
| Carbon dioxide (un–ionized) | Aqueous | CO2(aq) | −419.26 |
| Bicarbonate ion | Aqueous | HCO3– | −689.93 |
| Carbonate ion | Aqueous | CO32– | −675.23 |
| Monatomic chlorine | Gas | Cl | 121.7 |
| Chloride ion | Aqueous | Cl− | −167.2 |
| Chlorine | Gas | Cl2 | 0 |
| Chromium | Solid | Cr | 0 |
| Copper | Solid | Cu | 0 |
| Copper(II) oxide | Solid | CuO | −155.2 |
| Copper(II) sulfate | Aqueous | CuSO4 | −769.98 |
| Fluorine | Gas | F2 | 0 |
| Monatomic hydrogen | Gas | H | 218 |
| Hydrogen | Gas | H2 | 0 |
| Water | Gas | H2O | −241.818 |
| Water | Liquid | H2O | −285.8 |
| Hydrogen ion | Aqueous | H+ | 0 |
| Hydroxide ion | Aqueous | OH− | −230 |
| Hydrogen peroxide | Liquid | H2O2 | −187.8 |
| Phosphoric acid | Liquid | H3PO4 | −1288 |
| Hydrogen cyanide | Gas | HCN | 130.5 |
| Hydrogen bromide | Liquid | HBr | −36.3 |
| Hydrogen chloride | Gas | HCl | −92.30 |
| Hydrogen chloride | Aqueous | HCl | −167.2 |
| Hydrogen fluoride | Gas | HF | −273.3 |
| Hydrogen iodide | Gas | HI | 26.5 |
| Iodine | Solid | I2 | 0 |
| Iodine | Gas | I2 | 62.438 |
| Iodine | Aqueous | I2 | 23 |
| Iodide ion | Aqueous | I− | −55 |
| Iron | Solid | Fe | 0 |
| Iron carbide (Cementite) | Solid | Fe3C | 5.4 |
| Iron(II) carbonate (Siderite) | Solid | FeCO3 | −750.6 |
| Iron(III) chloride | Solid | FeCl3 | −399.4 |
| Iron(II) oxide (Wüstite) | Solid | FeO | −272 |
| Iron(II,III) oxide (Magnetite) | Solid | Fe3O4 | −1118.4 |
| Iron(III) oxide (Hematite) | Solid | Fe2O3 | −824.2 |
| Iron(II) sulfate | Solid | FeSO4 | −929 |
| Iron(III) sulfate | Solid | Fe2(SO4)3 | −2583 |
| Iron(II) sulfide | Solid | FeS | −102 |
| Pyrite | Solid | FeS2 | −178 |
| Lead | Solid | Pb | 0 |
| Lead dioxide | Solid | PbO2 | −277 |
| Lead sulfide | Solid | PbS | −100 |
| Lead sulfate | Solid | PbSO4 | −920 |
| Lead(II) nitrate | Solid | Pb(NO3)2 | −452 |
| Lead(II) sulfate | Solid | PbSO4 | −920 |
| Lithium fluoride | Solid | LiF | −616.93 |
| Magnesium | Solid | Mg | 0 |
| Magnesium ion | Aqueous | Mg2+ | −466.85 |
| Magnesium carbonate | Solid | MgCO3 | −1095.797 |
| Magnesium chloride | Solid | MgCl2 | −641.8 |
| Magnesium hydroxide | Solid | Mg(OH)2 | −924.54 |
| Magnesium hydroxide | Aqueous | Mg(OH)2 | −926.8 |
| Magnesium oxide | Solid | MgO | −601.6 |
| Magnesium sulfate | Solid | MgSO4 | −1278.2 |
| Manganese | Solid | Mn | 0 |
| Manganese(II) oxide | Solid | MnO | −384.9 |
| Manganese(IV) oxide | Solid | MnO2 | −519.7 |
| Manganese(III) oxide | Solid | Mn2O3 | −971 |
| Manganese(II,III) oxide | Solid | Mn3O4 | −1387 |
| Permanganate | Aqueous | MnO− 4 |
−543 |
| Mercury(II) oxide (red) | Solid | HgO | −90.83 |
| Mercury sulfide (red, cinnabar) | Solid | HgS | −58.2 |
| Nitrogen | Gas | N2 | 0 |
| Ammonia (ammonium hydroxide) | Aqueous | NH3 (NH4OH) | −80.8 |
| Ammonia | Gas | NH3 | −46.1 |
| Ammonium nitrate | Solid | NH4NO3 | −365.6 |
| Ammonium chloride | Solid | NH4Cl | −314.55 |
| Nitrogen dioxide | Gas | NO2 | 33.2 |
| Hydrazine | Gas | N2H4 | 95.4 |
| Hydrazine | Liquid | N2H4 | 50.6 |
| Nitrous oxide | Gas | N2O | 82.05 |
| Nitric oxide | Gas | NO | 90.29 |
| Dinitrogen tetroxide | Gas | N2O4 | 9.16 |
| Dinitrogen pentoxide | Solid | N2O5 | −43.1 |
| Dinitrogen pentoxide | Gas | N2O5 | 11.3 |
| Nitric acid | Aqueous | HNO3 | −207 |
| Monatomic oxygen | Gas | O | 249 |
| Oxygen | Gas | O2 | 0 |
| Ozone | Gas | O3 | 143 |
| White phosphorus | Solid | P4 | 0 |
| Red phosphorus | Solid | P | −17.4[5] |
| Black phosphorus | Solid | P | −39.3[5] |
| Phosphorus trichloride | Liquid | PCl3 | −319.7 |
| Phosphorus trichloride | Gas | PCl3 | −278 |
| Phosphorus pentachloride | Solid | PCl5 | −440 |
| Phosphorus pentachloride | Gas | PCl5 | −321 |
| Phosphorus pentoxide | Solid | P2O5 | −1505.5[6] |
| Potassium bromide | Solid | KBr | −392.2 |
| Potassium carbonate | Solid | K2CO3 | −1150 |
| Potassium chlorate | Solid | KClO3 | −391.4 |
| Potassium chloride | Solid | KCl | −436.68 |
| Potassium fluoride | Solid | KF | −562.6 |
| Potassium oxide | Solid | K2O | −363 |
| Potassium nitrate | Solid | KNO3 | −494.5 |
| Potassium perchlorate | Solid | KClO4 | −430.12 |
| Silicon | Gas | Si | 368.2 |
| Silicon carbide | Solid | SiC | −74.4,[7] −71.5[8] |
| Silicon tetrachloride | Liquid | SiCl4 | −640.1 |
| Silica (Quartz) | Solid | SiO2 | −910.86 |
| Silver bromide | Solid | AgBr | −99.5 |
| Silver chloride | Solid | AgCl | −127.01 |
| Silver iodide | Solid | AgI | −62.4 |
| Silver oxide | Solid | Ag2O | −31.1 |
| Silver sulfide | Solid | Ag2S | −31.8 |
| Sodium | Solid | Na | 0 |
| Sodium | Gas | Na | 107.5 |
| Sodium bicarbonate | Solid | NaHCO3 | −950.8 |
| Sodium carbonate | Solid | Na2CO3 | −1130.77 |
| Sodium chloride | Aqueous | NaCl | −407.27 |
| Sodium chloride | Solid | NaCl | −411.12 |
| Sodium chloride | Liquid | NaCl | −385.92 |
| Sodium chloride | Gas | NaCl | −181.42 |
| Sodium chlorate | Solid | NaClO3 | −365.4 |
| Sodium fluoride | Solid | NaF | −569.0 |
| Sodium hydroxide | Aqueous | NaOH | −469.15 |
| Sodium hydroxide | Solid | NaOH | −425.93 |
| Sodium hypochlorite | Solid | NaOCl | −347.1 |
| Sodium nitrate | Aqueous | NaNO3 | −446.2 |
| Sodium nitrate | Solid | NaNO3 | −424.8 |
| Sodium oxide | Solid | Na2O | −414.2 |
| Sulfur (monoclinic) | Solid | S8 | 0.3 |
| Sulfur (rhombic) | Solid | S8 | 0 |
| Hydrogen sulfide | Gas | H2S | −20.63 |
| Sulfur dioxide | Gas | SO2 | −296.84 |
| Sulfur trioxide | Gas | SO3 | −395.7 |
| Sulfuric acid | Liquid | H2SO4 | −814 |
| Titanium | Gas | Ti | 468 |
| Titanium tetrachloride | Gas | TiCl4 | −763.2 |
| Titanium tetrachloride | Liquid | TiCl4 | −804.2 |
| Titanium dioxide | Solid | TiO2 | −944.7 |
| Zinc | Gas | Zn | 130.7 |
| Zinc chloride | Solid | ZnCl2 | −415.1 |
| Zinc oxide | Solid | ZnO | −348.0 |
| Zinc sulfate | Solid | ZnSO4 | −980.14 |
Aliphatic hydrocarbons[edit]
| Formula | Name | ΔfH⦵ /(kcal/mol) | ΔfH⦵ /(kJ/mol) |
|---|---|---|---|
| Straight-chain | |||
| CH4 | Methane | −17.9 | −74.9 |
| C2H6 | Ethane | −20.0 | −83.7 |
| C2H4 | Ethylene | 12.5 | 52.5 |
| C2H2 | Acetylene | 54.2 | 226.8 |
| C3H8 | Propane | −25.0 | −104.6 |
| C4H10 | n-Butane | −30.0 | −125.5 |
| C5H12 | n-Pentane | −35.1 | −146.9 |
| C6H14 | n-Hexane | −40.0 | −167.4 |
| C7H16 | n-Heptane | −44.9 | −187.9 |
| C8H18 | n-Octane | −49.8 | −208.4 |
| C9H20 | n-Nonane | −54.8 | −229.3 |
| C10H22 | n-Decane | −59.6 | −249.4 |
| C4 Alkane branched isomers | |||
| C4H10 | Isobutane (methylpropane) | −32.1 | −134.3 |
| C5 Alkane branched isomers | |||
| C5H12 | Neopentane (dimethylpropane) | −40.1 | −167.8 |
| C5H12 | Isopentane (methylbutane) | −36.9 | −154.4 |
| C6 Alkane branched isomers | |||
| C6H14 | 2,2-Dimethylbutane | −44.5 | −186.2 |
| C6H14 | 2,3-Dimethylbutane | −42.5 | −177.8 |
| C6H14 | 2-Methylpentane (isohexane) | −41.8 | −174.9 |
| C6H14 | 3-Methylpentane | −41.1 | −172.0 |
| C7 Alkane branched isomers | |||
| C7H16 | 2,2-Dimethylpentane | −49.2 | −205.9 |
| C7H16 | 2,2,3-Trimethylbutane | −49.0 | −205.0 |
| C7H16 | 3,3-Dimethylpentane | −48.1 | −201.3 |
| C7H16 | 2,3-Dimethylpentane | −47.3 | −197.9 |
| C7H16 | 2,4-Dimethylpentane | −48.2 | −201.7 |
| C7H16 | 2-Methylhexane | −46.5 | −194.6 |
| C7H16 | 3-Methylhexane | −45.7 | −191.2 |
| C7H16 | 3-Ethylpentane | −45.3 | −189.5 |
| C8 Alkane branched isomers | |||
| C8H18 | 2,3-Dimethylhexane | −55.1 | −230.5 |
| C8H18 | 2,2,3,3-Tetramethylbutane | −53.9 | −225.5 |
| C8H18 | 2,2-Dimethylhexane | −53.7 | −224.7 |
| C8H18 | 2,2,4-Trimethylpentane (isooctane) | −53.5 | −223.8 |
| C8H18 | 2,5-Dimethylhexane | −53.2 | −222.6 |
| C8H18 | 2,2,3-Trimethylpentane | −52.6 | −220.1 |
| C8H18 | 3,3-Dimethylhexane | −52.6 | −220.1 |
| C8H18 | 2,4-Dimethylhexane | −52.4 | −219.2 |
| C8H18 | 2,3,4-Trimethylpentane | −51.9 | −217.1 |
| C8H18 | 2,3,3-Trimethylpentane | −51.7 | −216.3 |
| C8H18 | 2-Methylheptane | −51.5 | −215.5 |
| C8H18 | 3-Ethyl-3-Methylpentane | −51.4 | −215.1 |
| C8H18 | 3,4-Dimethylhexane | −50.9 | −213.0 |
| C8H18 | 3-Ethyl-2-Methylpentane | −50.4 | −210.9 |
| C8H18 | 3-Methylheptane | −60.3 | −252.5 |
| C8H18 | 4-Methylheptane | ? | ? |
| C8H18 | 3-Ethylhexane | ? | ? |
| C9 Alkane branched isomers (selected) | |||
| C9H20 | 2,2,4,4-Tetramethylpentane | −57.8 | −241.8 |
| C9H20 | 2,2,3,3-Tetramethylpentane | −56.7 | −237.2 |
| C9H20 | 2,2,3,4-Tetramethylpentane | −56.6 | −236.8 |
| C9H20 | 2,3,3,4-Tetramethylpentane | −56.4 | −236.0 |
| C9H20 | 3,3-Diethylpentane | −55.7 | −233.0 |
Other organic compounds[edit]
| Species | Phase | Chemical formula | ΔfH⦵ /(kJ/mol) |
|---|---|---|---|
| Acetone | Liquid | C3H6O | −248.4 |
| Benzene | Liquid | C6H6 | 48.95 |
| Benzoic acid | Solid | C7H6O2 | −385.2 |
| Carbon tetrachloride | Liquid | CCl4 | −135.4 |
| Carbon tetrachloride | Gas | CCl4 | −95.98 |
| Ethanol | Liquid | C2H5OH | −277.0 |
| Ethanol | Gas | C2H5OH | −235.3 |
| Glucose | Solid | C6H12O6 | −1271 |
| Isopropanol | Gas | C3H7OH | −318.1 |
| Methanol (methyl alcohol) | Liquid | CH3OH | −238.4 |
| Methanol (methyl alcohol) | Gas | CH3OH | −201.0 |
| Methyl linoleate (Biodiesel) | Gas | C19H34O2 | −356.3 |
| Sucrose | Solid | C12H22O11 | −2226.1 |
| Trichloromethane (Chloroform) | Liquid | CHCl3 | −134.47 |
| Trichloromethane (Chloroform) | Gas | CHCl3 | −103.18 |
| Vinyl chloride | Solid | C2H3Cl | −94.12 |
See also[edit]
- Calorimetry
- Thermochemistry
References[edit]
- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”) (1997). Online corrected version: (2006–) “standard pressure”. doi:10.1351/goldbook.S05921
- ^ Oxtoby, David W; Pat Gillis, H; Campion, Alan (2011). Principles of Modern Chemistry. p. 547. ISBN 978-0-8400-4931-5.
- ^ Moore, Stanitski, and Jurs. Chemistry: The Molecular Science. 3rd edition. 2008. ISBN 0-495-10521-X. pages 320–321.
- ^ “Enthalpies of Reaction”. www.science.uwaterloo.ca. Archived from the original on 25 October 2017. Retrieved 2 May 2018.
- ^ a b Housecroft, C. E.; Sharpe, A. G. (2004). Inorganic Chemistry (2nd ed.). Prentice Hall. p. 392. ISBN 978-0-13-039913-7.
- ^ Green, D.W., ed. (2007). Perry’s Chemical Engineers’ Handbook (8th ed.). Mcgraw-Hill. p. 2–191. ISBN 9780071422949.
- ^ Kleykamp, H. (1998). “Gibbs Energy of Formation of SiC: A contribution to the Thermodynamic Stability of the Modifications”. Berichte der Bunsengesellschaft für physikalische Chemie. 102 (9): 1231–1234. doi:10.1002/bbpc.19981020928.
- ^ “Silicon Carbide, Alpha (SiC)”. March 1967. Retrieved 5 February 2019.
- Zumdahl, Steven (2009). Chemical Principles (6th ed.). Boston. New York: Houghton Mifflin. pp. 384–387. ISBN 978-0-547-19626-8.
External links[edit]
- NIST Chemistry WebBook
