Глава VIII. ДИАГРАММА ТЕМПЕРАТУРА – ЭНТРОПИЯ
§ 39. Свойства системы координат температура — энтропия (Т — s)
При введении понятия энтропии было установлено, что эта величина является параметром состояния. Из уравнения
![]()

следует, что использовать энтропию s как параметр, характеризующий состояние рабочего тела, удобнее всего в сочетании с абсолютной температурой Т.
Если энтропию и температуру рассматривать как координаты некоторой плоскостной системы координат (рис. 42), то каждому сочетанию значений s и Т на плоскости координат (точки А, В и т. д.) будут соответствовать вполне определенные состояния рабочего тела. Очевидно также, что каждый процесс в координатах Т—s представляется некоторой линией, для которой в простейших случаях можно установить аналитическое выражение зависимости s = f (Т).
При нанесении какого-либо процесса на координатную плоскость sT необходимо знать абсолютное значение энтропии. Это значение, вообще говоря, может быть вычислено. Но для решения термодинамических задач в подавляющем большинстве случаев требуется определять только изменение энтропии в процессе. Поэтому вместо сложного вычисления абсолютных значений энтропии выбирают условный отсчет ее от какого-либо фиксированного состояния. Так, для идеального газа за начало отсчета энтропии принимают обычно нормальные физические условия (0° С и 760 мм рт. ст.), при которых энтропия условно считается равной нулю.
93
![]()
При таком способе отсчета энтропии изменение ее в процессах останется тем же, что и разность абсолютных значений энтропии.
Изменение энтропии в обратимом процессе в этом случае может быть подсчитано также по общей формуле
![]()
Если взять на кривой процесса А В (рис. 42) элементарный отрезок, в котором изменение температуры будет бесконечно мало (dT), то площадь под этим элементарным процессом (заштрихованная площадка) будет равна произведению текущего значения температуры Т на бесконечно малое приращение энтропии ds. Но, как известно, это произведение для обратимого процесса равно теплоте в обратимом процессе, т. е. dq = Tds.
Взяв бесконечно большую сумму бесконечно малых площадок по всему процессу АВ, получим площадь под кривой этого процесса. Аналитически эта сумма представляет собой интеграл от выражения Т ds в пределах от А до В, Этот интеграл численно равен теплоте в процессе, т. е.
![]()
Следовательно, в координатах Т—s теплота представляется площадью под линией процесса. Поэтому часто диаграмму Т—s называют тепловой диаграммой.
По характеру протекания линии процесса в координатах Т—s легко определить знак теплоты в процессе.
Из соотношения dq следует, что так как температура всегда положительна, знак dq определяется знаком ds. Если энтропия в процессе возрастает, то это значит, что теплота положительная, т. е. подводится, и наоборот. Аналитически это можно записать так: ds > 0 и dq > 0, ds < 0 и dq < 0.
Следует подчеркнуть, что в системе Т—s изображают процессы изменения состояния рабочего тела, и поэтому изменение энтропии относится только к рабочему телу, а не к системе, в которой этот процесс осуществляется.
![]()
следовательно, работа за цикл в тепловых единицах (дж) равна
Круговые_процессы или циклы в координатах Т—s, так же как и в любой другой системе координат, где по осям отложены параметры состояния, представляются замкнутыми линиями. Так как в таких процессах изменение внутренней энергии тела u = 0, то работа за цикл определяется как алгебраическая сумма подводимой и отводимой теплоты:
95

разности площадей под верхней и нижней ветвями цикла, т.е. I — пл. а1А2b — пл. а1В2b (рис. 43). Эта работа соответствует площади, ограниченной линиями цикла (пл. 1А2В1). Если площадь под линией процесса подвода теплоты больше площади, соответствующей отводимой теплоте, то работа цикла положительная, цикл 1А2В1 прямой. Для обратного цикла площадь, характеризующая подведенную теплоту, будет меньше площади, представляющей собой отведенную теплоту. Значит, работа за цикл отрицательная.
Очевидно, что термический к. п. д. цикла будет равен
![]()

Координаты Т—s с нанесенными на них графиками простейших процессов для данного рабочего тела называют диаграммой T—s этого тела. Простейшей тепловой диаграммой будет диаграмма для идеального газа. Для построения этих диаграмм необходимо рассмотреть основные процессы идеального газа в координатах Т—s.
§ 40. Основные процессы идеального газа в координатах Т—s
![]()
Из этого следует, что
![]()
Для процесса v — const (рис. 44) изменение энтропии в пределах от точки 1 до точки 2 выразится как
![]()
Считая, что теплоемкость в процессе остается постоянной, получаем
Изохорный процесс (v = const). Для изохорного процесса бесконечно малое количество теплоты
![]()
![]()

или, переходя к десятичным логарифмам,
![]()
Изохора в координатах Т—s представляется логарифмической кривой 1—2. Площадь под линией процесса а12b соответствует теплоте в процессе
![]()
которая равна изменению внутренней энергии рабочего тела (работа в процессе равна нулю):
Такой же вывод получается при рассмотрении процессов
![]()

Принимая теплоемкость в процессе постоянной, получаем
![]()
или
![]()
Изобарный процесс, так же как и изохорный, в координатах Т—s представляется логарифмической кривой. Площадь под линией процесса а12b соответствует теплоте в процессе
![]()
эта теплота равна изменению энтальпии рабочего тела.
Рассмотрение изобары и изохоры, проходящих через одну и ту же точку (рис. 46), приводит к выводу, что изохора расположена круче, чем изобара. Это объясняется следующим образом:
![]()
Изотермический процесс (Т = const). В координатах этот процесс представляется прямой, параллельной оси абсцисс (рис. 47). Уравнение процесса: Т = const.

![]()
Для изотермического процесса идеального газа
![]()
изменение энтропии
![]()
Заменяя по уравнению состояния газа
![]()
получаем
97
![]()
![]()

Для процесса 1—2 (рис. 47)
или, переходя к десятичным логарифмам, получим
Отношение объемов можно заменить через обратное отношение давлений. Тогда
![]()
Для любых двух изохор v1 = const и v2 = const (рис. 48) легко установить, что расстояния между ними в направлении, параллельном оси абсцисс, одинаковы. Это следует из того, что для любой изо-

термы, расположенной между этими изохорами, изменение энтропии рабочего тела будет одно и то же:
![]()
Это значит, что все изохоры эквидистантны в направлении, параллельном оси абсцисс. Подобным же образом может быть показана эквидистантность изобар в том же направлении:
![]()
Адиабатный процесс (dq = 0)._ Для этого процесса, так же как
и для любого другого:
Теплота отводится от рабочего тела по изотерме cd. Количество отводимой теплоты соответствует площади
![]()
![]() 99
99
Для адиабатного процесса dq = 0, поэтому ds = 0. Отсюда следует, что обратимый адиабатный процесс характеризуется постоян-
ством энтропии s = const. На этом основании такой процесс часто называют изоэнтропным.
Очевидно, графиком такого процесса явится прямая, параллельная оси ординат (рис. 49). Такой вид графика адиабатного процесса указывает на то, что этот процесс характеризуется не только тем, что dq = 0, но и тем, что q = 0. Это находится в полном соответствии со всем сказанным выше.
§ 41. Диаграмма Т —s для идеального газа
На рис. 50 приведен пример общего вида части диаграммы для газа, подчиняющегося уравнению состояния газа. Сплошными горизонтальными и вертикальными линиями нанесены соответственно изотермы и адиабаты. Начало координат по оси энтропии (ось абсцисс) выбирается условно.
Сплошные логарифмические кривые представляют собой изобары. При этом изобары, соответствующие большим давлениям, располагаются ближе к оси ординат, т. е.
Рп > Р1
Пунктирными логарифмическими кривыми нанесены изохоры. Изохоры, расположенные ближе к оси ординат, соответствуют меньшим объемам, т. е. vn < v1.
Такой диаграммой удобно пользоваться при исследовании процессов и циклов, протекающих в рабочих телах, которые можно принимать за идеальные газы. Это допустимо, например, при исследовании рабочего процесса воздушных и некоторых газовых компрессоров, двигателей внутреннего сгорания, газотурбинных установок и т. п. В качестве примера в приложении дана диаграмма Т—s для воздуха, который принимается за идеальный газ.
§ 42. Цикл Карно на диаграмме Т — s
![]()
Цикл Карно, состоящий из двух изотерм и двух адиабат, представится в координатах T—s в виде прямоугольника abed (рис. 51). Изотерма ab соответствует процессу подвода теплоты. Количество подводимой к рабочему телу теплоты определяется как площадь
| Энтропия | |
|---|---|
 |
|
| Размерность |
 |
| Единицы измерения | |
| СИ | Дж/К |
| СГС | эрг/К |
| Термодинамические величины |
|---|
 |
| Статья является частью серии «Термодинамика» |
См. также: Термодинамические потенциалы. |
| Разделы термодинамики |
|
| См. также «Физический портал» |
Термодинамическая энтропия 
Утверждение о существовании энтропии и перечисление её свойств составляют содержание второго и третьего начал термодинамики. Значимость данной величины для физики обусловлена тем, что наряду с температурой её используют для описания термических явлений и термических свойств макроскопических объектов. Качественные представления о термическом состоянии системы связаны с тепловыми ощущениями, выражаемыми понятиями «теплее», «холоднее», «нагрев», «охлаждение», «степень нагретости». К термическим относят свойства, характеризующие поведение вещества при его нагреве или охлаждении: термические коэффициенты, теплоёмкость и другие калорические коэффициенты, постоянную Кюри, показатели термостойкости, пределы огнестойкости и т. д.; примерами термических явлений служат термическое расширение, пироэлектричество, электрокалорический эффект, теплопроводность, изменение агрегатного состояния — кристаллизация и замерзание, плавление и таяние, испарение, кипение, сублимация (возгонка), конденсация и другие процессы.
Историческая справка[править | править код]
Понятие энтропии, её обозначение и название были введены Р. Клаузиусом (1865). Абстрактность этого понятия — одного из краеугольных камней термодинамики — и разнообразие подходов к обоснованию существования энтропии как термодинамической величины привели к появлению аксиоматики термодинамики.
Термодинамическая дефиниция энтропии[править | править код]
В термодинамике энтропию вводят, обосновывая её существование, перечисляя её свойства и строя для неё шкалу измерения на основании первого, второго и третьего начал термодинамики.
В термодинамических формализмах Клаузиуса[1] и Каратеодори[2] энтропию вводят одновременно с абсолютной термодинамической температурой. Математический аппарат термодинамики Гиббса[3] основан на использовании энтропии в качестве независимой термодинамической переменной, тогда как температура — естественный кандидат на эту роль, вводится как функция внутренней энергии и энтропии. Наконец, в рациональной термодинамике энтропию выражают через внутреннюю энергию и температуру, которые рассматривают как основные неопределяемые переменные теории.
Энтропия простой системы[править | править код]
Первое начало (закон) термодинамики устанавливает связь между внутренней энергией, работой и теплотой: одна из этих физических величин задаётся с помощью двух других, которые, будучи исходными объектами теории, в рамках самой этой теории определены быть не могут просто потому, что не существует понятий более общих, под которые их можно было бы подвести[4]. Термодинамика заимствует понятия энергии и работы из других разделов физики[5][6], тогда как определение количеству теплоты, наоборот, даётся только и именно в термодинамике. Согласно Клаузиусу теплоту 



|
(Дефиниция теплоты по Клаузиусу) |
Первое начало в этой формулировке вводит теплоту как физическую характеристику процесса, поведение которой определяется законом сохранения энергии, но не определяет её как математический объект. Детализировать дефиницию теплоты проще всего для равновесного процесса, когда работу, а следовательно и теплоту, можно выразить через переменные состояния. Для бесконечно малого[11] равновесного процесса в простой системе[12] возможен единственный вид работы — работа расширения/сжатия 

|
(Работа расширения/сжатия для равновесного процесса в простой системе) |
где 



|
(Первое начало для равновесного процесса в простой системе) |
где 
Это выражение, определяющее элементарную теплоту как математический объект, есть линейная дифференциальная форма (форма Пфаффа) для двух независимых переменных. Для данной пфаффовой формы условие интегрируемости Эйлера не выполняется, то есть 


|
(Дефиниция энтропии простой равновесной системы) |
где

есть интегрирующий делитель для формы Пфаффа. Клаузиус назвал функцию состояния 
Интегрирование уравнения для энтропии приводит к появлению в выражении для функции
Энтропия закрытой системы в термодинамике Клаузиуса — Каратеодори[править | править код]
Традиционный подход к построению термодинамики (аксиоматика Клаузиуса — Каратеодори) основан на использовании представления о внутренней энергии как базовом понятии теории, заимствовании формул для вычисления термодинамической работы из механики и электродинамики сплошных сред, и первом начале термодинамики в формулировке Клаузиуса.
Помимо работы расширения/сжатия система может одновременно выполнять другие виды работ, например работу по изменению площади поверхности раздела фаз, работу перемещения в поле тяготения, работу поляризации диэлектрика в электрическом поле и т. д. Объединяет все эти виды работ формальная структурная идентичность расчётных формул друг с другом и с выражением для работы расширения/сжатия[23][24][25]:

|
где 



Если однородная система одновременно совершает несколько различных видов работ, то они суммируются и полная работа системы 

|
(Суммарная работа для равновесного процесса в однородной системе) |
а для первого начала термодинамики получаем соотношение[30][31]:

|
(Первое начало для равновесного процесса в однородной системе) |
которое как и в случае простой системы представляет собой форму Пфаффа. Следствием второго начала термодинамики в любой его формулировке является вывод о том, что пфаффова форма

|
(Энтропия однородной закрытой системы) |
и абсолютная термодинамическая температура

Выражение 

Энтропия открытой системы[править | править код]
Принимая, что теплота и работа являются двумя единственно возможными формами передачи энергии[32][33][34], а изменение энергии, связанное с переносом вещества в открытой системе, есть составная часть общей работы, называемая химической работой (работой перераспределения масс веществ[35]), в случае однородной открытой системы дифференциальную форму 

|
(Химическая работа для равновесного процесса в открытой однородной системе) |
где 


Пфаффова форма

|
(Первое начало для равновесного процесса в открытой однородной системе) |
Дальнейшие рассуждения о существовании энтропии
и абсолютной термодинамической температуры

для открытой системы ничем не отличаются от соображений, высказанных при рассмотрении закрытой системы, поэтому ниже перечислены причины, по которым открытые системы потребовали отдельного рассмотрения.
Первая из этих причин состоит в том, что использование в понятийном аппарате термодинамики химической работы как части общей работы делает неэквивалентными представления об адиабатной изоляции как накладывающей запрет на обмен веществом (то есть любая адиабатно изолированная система есть система закрытая или, говоря иначе, масса есть адиабатически заторможенная величина)[42][43][44][45], и адиабатной изоляции как допускающей обмен энергией только в форме работы[46][47]. Восстановить эквивалентность приведённых выше формулировок об адиабатной изоляции удаётся, если модифицировать дефиницию теплоты по Клаузиусу, добавив к теплоте и работе третью форму передачи энергии — энергию переноса массы 

|
(Модифицированная дефиниция теплоты для открытой системы) |
где
Вторая причина отдельного рассмотрения вопроса об энтропии открытых систем заключается в следующем. За исключением химического потенциала все входящие в пфаффову форму

|
(Энтропия открытой однородной системы по Гиббсу; фундаментальное уравнение Гиббса в энтропийном выражении) |
как функции состояния, при неизменности масс компонентов совпадающей с энтропией однородной закрытой системы. Из фундаментального уравнения Гиббса в дифференциальной форме[55]

|
(Дифференциальная форма фундаментального уравнения Гиббса для энтропии) |
находим значения частных производных энтропии:



Химический потенциал 

|
(Дефиниция химического потенциала компонента) |
Энтропия в термодинамике Гиббса[править | править код]
Построение теории на основе постулирования существования энтропии как функции состояния, в состав независимых переменных которой входят массы компонентов, составляет главное содержание термодинамики Гиббса[57], а способ, каким выполнено распространение термодинамики Клаузиуса на открытые системы, позволяет говорить об аксиоматике Гиббса[52][45]. В термодинамике Гиббса вводят понятия компонента системы, фазы и многофазной гетерогенной системы, постулируют существование внутренней энергии 
Обратите внимание, что содержательную дефиницию температуры по Гиббсу[59][60][61]

|
(Термодинамическая температура по Гиббсу) |
можно, с другой стороны, рассматривать и как описательную дефиницию энтропии. А именно, энтропия в термодинамике Гиббса есть такая экстенсивная переменная состояния, что производная внутренней энергии по энтропии представляет собой интенсивную переменную состояния, обладающую всеми положенными термодинамической температуре свойствами.
Энтропия в рациональной термодинамике[править | править код]
Рациональная термодинамика не подразделяет термодинамику на равновесную и неравновесную; обе эти дисциплины рассматриваются как единая часть физики сплошных сред[62][63][64][65]. Равновесная рациональная термодинамика есть результат применения общей теории к системам в состоянии равновесия[66]. Исходные неопределяемые понятия теории — энергия 


при механическом равновесии имеющем одинаковое значение во всех частях системы, вводят химический потенциал

как интенсивную величину, имеющую при химическом равновесии одно и то же значение во всех частях системы[67].
Абсолютную термодинамическую температуру вводят посредством следующей аксиомы: существует интенсивная термодинамическая величина, температура

которая характеризует степень нагретости тел и обладает следующими свойствами[68]:
- в выбранном за начало отсчёта состоянии температура равна нулю
;
- температура монотонно растёт с увеличением энергии системы
;
- при термодинамическом равновесии имеет одно и то же значение во всех частях системы.
Энтропию в рациональной термодинамике задают как аддитивную величину, равную[69]

Свойства энтропии, вытекающие из этого определения[69]:
;
;
;
где 
Свойства энтропии[править | править код]
Перечисление свойств энтропии дано применительно к термодинамике Гиббса; примеры, приводимые для иллюстрации перечисляемых свойств энтропии, относятся, как правило, к открытым однородным термодеформационным системам, для которых справедливо фундаментальное уравнение Гиббса в энтропийном выражении[70][71]:

|
(Фундаментальное уравнение Гиббса в энтропийном выражении для открытой термодеформационной системы) |
- Энтропия в фундаментальном уравнении в энтропийном выражении есть однозначная дифференцируемая функция аддитивных независимых переменных[72][44][73] с непрерывными первыми и кусочно-непрерывными вторыми производными.
- Энтропия есть величина аддитивная[74][75][76][72][77][78][79][80], то есть энтропия термодинамической системы равна сумме энтропий всех её частей. Аддитивность энтропии позволяет распространить это понятие на термодинамические системы любой сложности.
- Как следствие аддитивности получаем, что энтропия в фундаментальном уравнении в энтропийном выражении есть однородная функция первого порядка всех независимых переменных[81][73], то есть для


|
- и для неё справедливо тождество (теорема) Эйлера[82]:

|
- Для однородной системы частная производная энтропии по внутренней энергии есть величина, обратная абсолютной термодинамической температуре (термодинамическая дефиниция температуры как следствие второго начала термодинамики)[83][60][61][84]:
![Tequiv left[left({frac {partial S}{partial U}}right)_{{V,{x_{i}}}}right]^{{-1}}.](https://wikimedia.org/api/rest_v1/media/math/render/svg/0befaea6b14739d02ea258e6fd840d6e6790518b)
|
(Термодинамическая дефиниция температуры) |
- В соответствии с теоремой об обратных величинах[85] это определение совпадает с дефиницией равновесной температурой по Гиббсу[59][60][61]:

|
(Термодинамическая температура по Гиббсу) |
- Энтропия есть монотонная функция внутренней энергии
- Энтропия есть величина полуограниченная. Традиционно принимают, что энтропия ограничена снизу, то есть для каждой термодинамической системы существует состояние с наименьшей энтропией[88].
- Температура есть величина полуограниченная. Абсолютная температурная шкала Кельвина построена так, что

- Нуль и бесконечность допустимы в качестве пределов[88].
- Для любой термодинамической системы состояния с наименьшей энтропией и наименьшей температурой совпадают (постулат Планка)[88][89]. С приближением температуры к абсолютному нулю энтропия перестаёт зависеть от температуры и приближается к определённому постоянному значению, которое можно положить равным нулю[90] и принять за начало отсчёта энтропии, устранив тем самым упомянутый в разделе Энтропия простой системы произвол в выборе постоянной интегрирования для энтропии:

|
(Третье начало термодинамики; тепловая теорема Нернста) |
- Согласно постулату Тиссы внутренняя энергия ограничена и эта граница соответствует абсолютному нулю температуры[88][91]. Таким образом, состояние системы при абсолютном нуле температуры, когда все термодинамические величины, характеризующие равновесное состояние, перестают зависеть от температуры[92], наилучшим образом подходит в качестве стандартного состояния начала отсчёта основных термодинамических величин.
- Энтропия изолированной системы в состоянии термодинамического равновесия имеет максимальное значение (постулат Гиббса)[93][92], то есть для равновесия изолированной системы необходимо и достаточно, чтобы при всех возможных (не нарушающих постоянства внутренней энергии, обобщённых координат и масс компонентов) изменениях её состояния вариация энтропии
системы не была положительной[94]:

|
(Условие равновесия изолированной системы) |
- Поскольку речь идёт об изолированной системе, внешнее воздействие на которую запрещено, понятие вариации в данном случае означает виртуальное изменение энтропии[95]. Знак равенства в этом выражении относится к безразличному равновесию.
- Условие равновесия Гиббса вытекает из входящего в состав второго начала термодинамики постулата Клаузиуса о неубывании энтропии адиабатно изолированной системы[94]
- Энтропия в фундаментальном уравнении в энтропийном выражении есть характеристическая функция, то есть фундаментальное уравнение содержит все термодинамические сведения о данной системе[73].
Энтропия как характеристическая функция[править | править код]
Энтропия и теплота квазистатического (равновесного) процесса[править | править код]
Из выражения для первого начала в открытой однородной системе и дифференциальной формы фундаментального уравнения Гиббса для энтропии получаем выражение для элементарной теплоты равновесного (квазистатического) процесса[96][97]:

|
(Теплота элементарного равновесного процесса) |
(для простой равновесной системы это выражение непосредственно вытекает из дефиниции энтропии).
Данное соотношение, связывающее термодинамику Клаузиуса с термодинамикой Гиббса, представляет интерес для пользователей, которым требуется изложить материал из старой учебной и научной литературы с применением терминологии, либо вовсе не использующей понятие «теплота», либо использующей его как определяемое через энтропию и абсолютную температуру вторичное понятие.
Энтропия как характеристика изотермического процесса[править | править код]

Зависимость удельной энтропии воды от температуры
Для равновесного изотермического процесса с нулевой работой интегрирование выражения для теплоты равновесного процесса даёт следующее выражение для изменения внутренней энергии:

то есть в любом равновесном изотермическом процессе с нулевой работой энергия расходуется на увеличение энтропии системы и выделяется при уменьшении энтропии. Преобразуем это уравнение к виду

и назовём отношение 
Рассмотрим в качестве примера фазовые переходы в воде при атмосферном давлении (см. рисунок). При таянии льда подводимая к системе энергия расходуется на увеличение энтропии системы вследствие изменения структуры H2O, тогда как температура системы лёд + вода остаётся близкой к 0 °C (273 К) до полного исчезновения льда. При замерзании воды имеет место обратная ситуация: энергия выделяется в окружающую среду при 0 °C. Нагрев воды, образовавшейся при таянии льда, ведёт к повышению температуры воды вплоть до её закипания при 100 °C (373 К). Кипение воды при постоянном давлении есть процесс изотермический: подводимая энергия расходуется на испарение воды и увеличение энтропии системы вода + водяной пар, тогда как температура остаётся близкой к 100 °C до полного исчезновения жидкой воды.

Статистическое определение энтропии: принцип Больцмана[править | править код]
В 1877 году Людвиг Больцман установил связь энтропии с вероятностью данного состояния. Позднее эту связь представил в виде формулы Макс Планк:

где константа 

Рассмотрим, например, идеальный газ в сосуде. Микросостояние определено как позиции и импульсы (моменты движения) каждого составляющего систему атома. Связность предъявляет к нам требования рассматривать только те микросостояния, для которых:
(I) месторасположения всех частей расположены в рамках сосуда,
(II) для получения общей энергии газа кинетические энергии атомов суммируются.
Согласно определению, энтропия является функцией состояния, то есть не зависит от способа достижения
этого состояния, а определяется параметрами этого состояния. Так как
Понимание энтропии как меры беспорядка[править | править код]
Существует мнение, что мы можем смотреть на энтропию и как на меру беспорядка в системе. В определённом смысле это может быть оправдано, потому что мы думаем об «упорядоченных» системах как о системах, имеющих очень малую возможность конфигурирования, а о «беспорядочных» системах как об имеющих очень много возможных состояний. Собственно, это просто переформулированное определение энтропии как числа микросостояний на данное макросостояние.
Рассмотрим, например, распределение молекул идеального газа. В случае идеального газа наиболее вероятным состоянием, соответствующим максимуму энтропии, будет равномерное распределение молекул. При этом реализуется и максимальный «беспорядок», так как при этом будут максимальные возможности конфигурирования.
Получившее повсеместное распространение понимание энтропии как меры беспорядка в термодинамической системе не является, тем не менее, общепринятым[98]: «Тождественность энтропии с беспорядком не только никем никогда не была доказана и не только не может быть доказана в принципе, но и прямо противоречит реально наблюдаемым фактам…»[98]; «…применительно к реальным системам энтропия не является мерой беспорядка»[99]; «…в ходе роста энтропии Вселенной общая её (Вселенной) сложность растёт, однако для составляющих Вселенную реальных (под)систем энтропия мерой беспорядка/сложности не является»[100].
Границы применимости понимания энтропии как меры беспорядка[править | править код]
Подобное определение беспорядка термодинамической системы как количества возможностей конфигурирования системы фактически дословно соответствует определению энтропии как числа микросостояний на данное макросостояние. Проблемы начинаются в двух случаях:
- когда начинают смешивать различные понимания беспорядка, и энтропия становится мерой беспорядка вообще;
- когда понятие энтропии применяется для систем, не являющихся термодинамическими.
В обоих этих случаях применение понятия термодинамической энтропии совершенно неправомерно[101].
Рассмотрим оба пункта подробнее.
Рассмотрим пример термодинамической системы — распределение молекул в поле тяготения. В этом случае наиболее вероятным распределением молекул будет распределение согласно барометрической формуле Больцмана. Другой пример — учёт электромагнитных сил взаимодействия между ионами. В этом случае наиболее вероятным состоянием, соответствующим минимуму свободной энергии, будет упорядоченное кристаллическое состояние, а совсем не «хаос», хотя в состоянии «хаоса» значение конфигурационной энтропии системы и ниже. (Термин «хаос» здесь понимается в смысле беспорядка — в наивном смысле. К хаосу в математическом смысле как сильно неустойчивой нелинейной системе это не имеет отношения, конечно.)
Рассмотрим случай с кристаллической решёткой более подробно. Кристаллическая решётка может быть и в равновесном, и в неравновесном состоянии, как и любая термодинамическая система. Скажем, возьмём следующую модель — совокупность взаимодействующих осцилляторов. Рассмотрим некоторое неравновесное состояние: все осцилляторы имеют одинаковое отклонение от положения равновесия. С течением времени эта система перейдёт в состояние ТД равновесия, в котором отклонения (в каждый момент времени) будут подчинены некоторому распределению типа Максвелла (только это распределение будет для отклонений, и оно будет зависеть от типа взаимодействия осцилляторов). В таком случае максимум энтропии будет действительно реализовывать максимум возможностей конфигурирования, то есть — беспорядок согласно вышеуказанному определению. Но данный «беспорядок» вовсе не соответствует «беспорядку» в каком-либо другом понимании, например, информационному. Такая же ситуация возникает и в примере с кристаллизацией переохлаждённой жидкости, в которой образование структур из «хаотичной» жидкости идёт параллельно с увеличением энтропии.
То есть при образовании кристалла из переохлаждённой жидкости энтропия увеличивается с одновременным ростом температуры. Если кристаллизация сопровождается отводом тепла из системы, то энтропия при этом уменьшится.
Это неверное понимание энтропии появилось во время развития теории информации, в связи с парадоксом термодинамики, связанным с мысленным экспериментом так называемый «демона Максвелла». Суть парадокса заключалась в том, что рассматривалось два сосуда с разными температурами, соединённых узкой трубкой с затворками, которыми управлял так называемый «демон». «Демон» мог измерять скорость отдельных летящих молекул, и таким образом избирательно пропускать более быстрые в сосуд с высокой температурой, а более медленные — в сосуд с низкой. Из этого мысленного эксперимента вытекало кажущееся противоречие со вторым началом термодинамики.
Парадокс может быть разрешён при помощи теории информации. Для измерения скорости молекулы «демон» должен был бы получить информацию о её скорости. Но всякое получение информации — материальный процесс, сопровождающийся возрастанием энтропии. Количественный анализ[102] показал, что приращение энтропии при измерении превосходит по абсолютной величине уменьшение энтропии, вызванное перераспределением молекул «демоном».
Измерение энтропии[править | править код]
В реальных экспериментах очень трудно измерить энтропию системы. Техники измерения базируются на термодинамическом определении энтропии и требуют экстремально аккуратной калориметрии.
Для упрощения мы будем исследовать механическую систему, термодинамические состояния которой будут определены через её объём 

где нижний индекс 

Таким образом, мы можем получить значение энтропии любого состояния (


В добавление, если путь между первым и последним состояниями лежит сквозь любой фазовый переход первого рода, скрытая теплота, ассоциированная с переходом, должна также учитываться.
Энтропия первоначального состояния должна быть определена независимо. В идеальном варианте выбирается первоначальное состояние как состояние при экстремально высокой температуре, при которой система существует в виде газа. Энтропия в этом состоянии подобна энтропии классического идеального газа плюс взнос от молекулярных вращений и колебаний, которые могут быть определены спектроскопически.
Построение графика изменения энтропии[править | править код]
Следующее уравнение может быть использовано для построения графика изменения энтропии на диаграмме 

Здесь два замечания:
«Что такое энтропия?»[править | править код]

Что сложнее: паровоз или лошадь? Когда появились первые паровозы, путеец рассказал обратившимся к нему ошеломлённым крестьянам об устройстве и действии паровой машины и закончил объяснение вопросом: «Всё понятно?» — «Всё! Кроме того, где же внутри находится лошадь?» Крестьяне с детства знают лошадь, она для них в объяснении не нуждается. С научной точки зрения лошадь несравненно более сложна, чем тепловая машина, так что наука пыталась представить лошадь как тепловую машину, а не наоборот
Однозначного ответа на этот вопрос не существует по той простой причине, что разных энтропий много — представление об энтропии используется в различных научных дисциплинах: термодинамике, статистической физике, теории информации и др. Но и внутри каждой из перечисленных дисциплин единообразия также нет и в помине: в теории информации рассматривают энтропию Шеннона, энтропию Реньи, энтропию Чисара, энтропию Хаврда — Чарват — Дароши[103]; статистическая физика оперирует энтропиями Больцмана, Гиббса, Цаллиса; существуют различные дефиниции термодинамической энтропии. Содержательная дефиниция той или иной конкретной энтропии зависит от аксиоматики системы построения/изложения, использующей эту энтропию. По указанной причине не существует универсальной дефиниции термодинамической энтропии, ибо для различных аксиоматических систем термодинамики ответ на вынесенный в заголовок вопрос будет различен.
Студенты, приступающие к изучению термодинамики, часто жалуются на непонятность энтропии, связанную с отсутствием наглядности (рисунок иллюстрирует относительность представлений людей о наглядности, понятности и простоте термодинамических систем[K 1]).
Для описания термических явлений в физике вводят новые комплементарные (взаимодополняющие) физические величины — температуру и энтропию, — содержательные дефиниции которым не дают ни в механике, ни в электродинамике. В термодинамике Клаузиуса — Каратеодори энтропию вводят как приведённую внутреннюю энергию изотермической системы, то есть разность энтропий равна приведённой теплоте изотермического процесса.
В термодинамике Гиббса и в системе А. А. Гухмана энтропия представляет собой неопределяемое базовое понятие — таков в этих системах содержательный ответ на рассматриваемый вопрос[K 2]. В термодинамической аксиоматике А. А. Гухмана [105][106][107][108][109][110] и рациональной термодинамике в трактовке П. А. Жилина[65][K 3] и температуру, и энтропию вводят как неопределяемые базовые переменные, несводимые к более простым[K 4]. Пояснительное дополнение к дефиниции энтропии по Гухману разъясняет, что энтропия есть координата состояния при термическом взаимодействии (теплообмене), которое выражается в переносе энтропии от одного объекта к другому[112].
В рациональной термодинамике в трактовке школы Трусделла в качестве базовой термической переменной используют температуру

но, в отличие от другой функции энергии и температуры, — теплоёмкости 

в формулу, служащую дефиницией энтропии, входит не производная, а интеграл. Пояснительное дополнение к ответу, пригодное почти[K 5] для любого способа изложения термодинамики, сообщает, что энтропия необходима для построения математического аппарата термодинамики и, следовательно, привлечение термодинамики к решению любой научной или практической задачи явно или неявно подразумевает использование энтропии. Польза, приносимая людям от обращения к термодинамике, есть польза от ввода энтропии в понятийный аппарат науки. Дополнительно к сказанному можно провести аналогию с теплоёмкостью: если для неизотермических процессов нагрева льда, жидкой воды и водяного пара затраты энергии равны произведению теплоёмкости на разность температур, то для изотермических процессов таяния и кипения затраты энергии равны произведению температуры на разность энтропий.
Понять энтропию и её значение для термодинамики означает, что необходимо знать происхождение этой величины, понимать её связи с другими термодинамическими переменными и уметь применять энтропию на практике[K 6][104].
См. также[править | править код]
- Аксиоматика термодинамики
- Внутренняя энергия
- Демон Максвелла
- Негэнтропия
- Стрела времени
- Тепловая смерть
- Температура
- Термодинамика чёрных дыр
- Характеристическая функция
- Энтропия
- Энтропия Гиббса
- Энтропия Реньи
- Энтропия Цаллиса
- Энтропия Чисара
- Энтропия Шеннона
Комментарии[править | править код]
- ↑ Наглядность, понятность, очевидность и простота есть суждения относительные, зависящее как от обыденности понятия, так и от уровня знаний человека. Крестьяне с детства знали лошадь, и она для них наглядна и понятна. Для теплотехников наглядна и понятна тепловая машина, а не лошадь. В. Томсон как-то на лекции спросил студентов: «Знаете ли вы, кто такой математик?» Написав на аудиторной доске:
, Томсон повернулся к студентам и, указывая на эту формулу, сказал: «Математик — тот, для кого это так же очевидно, как для вас то, что дважды два — четыре»[104].
- ↑ Описательная характеристика энтропии как термической (тепловой) координаты состояния не отменяет того факта, что в системе Гухмана энтропия входит в число основных неопределяемых понятий теории.
- ↑ Вот цитата из статьи К. Трусделла, демонстрирующая совпадение его взглядов с подходом П. А. Жилина: «Я повторяю в течение уже многих лет, пренебрегая насмешками людей, наделённых физической интуицией, что температура и энтропия являются наряду с массой, положением и временем первоначальными неопределяемыми переменными. Они описываются только такими свойствами, которые можно выразить языком математики»[111]. Эти воззрения на температуру и энтропию отличаются от тех, которые сейчас принято рассматривать как отличительную особенность «рациональной термодинамике в трактовке школы Трусделла».
- ↑ Чтобы дать содержательную дефиницию какому-либо понятию, нужно указать, частным случаем какого более общего понятия оно является. Если более фундаментального понятия не существует, то понятие в конце цепочки дефиниций является неопределяемым — базовым (первичным, исходным, начальным) понятием аксиоматической системы, несводимым к более простым. В любой науке имеются такие первичные понятия, те элементарные кирпичики, из которых строятся все остальные, производные понятия, и которым не даются содержательные дефиниции в самой научной дисциплине. Примерами неопределяемых базовых понятий служат: в математике — множество, в физике — пространство, время, масса, энергия и др. Невозможность дать понятию или переменной содержательной дефиниции без выхода за границы изучаемой дисциплины, во-первых, не означает запрета на использование для базового понятия/переменной описательных дефиниций, и, во-вторых, свойства базовых понятий/переменных описываются аксиомами рассматриваемой теории. Иными словами, набор базовых понятий/переменных научной дисциплины зависит от выбора системы изложения/построения этой дисциплины, а полный набор её аксиом образует систему содержательных дефиниций базовых понятий/переменных теории.
- ↑ Слово «почти» служит напоминанием о том, что любую систему построения/изложения термодинамики, в которой энтропия есть понятие вторичное (выводимое из понятий более общих), можно в принципе преобразовать в другую систему — «безэнтропийную термодинамику», — в которой энтропию как понятие необязательное уже не используют[115]. В связи со сказанным упомянем, что автор третьего закона термодинамики Вальтер Нернст в своих работах не «пользовался понятием энтропии, которое он считал неясным и потому попросту не любил его»[116] (Нернст использовал свободную энергию Гельмгольца, связанную с максимальной работой, то есть энтропию он заменял взятой со знаком «минус» производной от энергии Гельмгольца по температуре[117]).
- ↑ В связи со сказанным представляют интерес воспоминания И. К. Кикоина, посещавшего в студенческие годы семинар В. А. Фока и рассказавшего историю про поиск решения сложной задачи по электростатике: «…в конце концов, получили длиннющее дифференциальное уравнение. Оно занимало всю доску. За математическими выкладками мы следили очень внимательно, так что с математикой всё было в порядке, а вот усмотреть физический смысл, скрытый за этой длинной формулой, мы не могли. Кто-то из студентов спросил Владимира Александровича: “А какой физический смысл имеет это уравнение?”. — Он на нас посмотрел с укором и сказал: “А физический смысл этого уравнения заключается в том, что оно имеет решение”»[118].
Примечания[править | править код]
- ↑ Клаузиус, 1934.
- ↑ Каратеодори.
- ↑ Гиббс Дж. В., Термодинамика. Статистическая механика, 1982.
- ↑ Хазен, 2000, глава VI, раздел 4. Полная и замкнутая формулировка аксиоматического определения энтропии и начал термодинамики..
- ↑ Петров Н., Бранков Й., Современные проблемы термодинамики, 1986, с. 35.
- ↑ Семенченко, 1966, с. 54.
- ↑ Clausius, 1887, S. 33.
- ↑ Борн, 1964, с. 230–231.
- ↑ Знак
или
перед
- ↑ Борн, 1964, с. 231.
- ↑ Элементарным (инфинитезимальным) называют процесс, для которого разница между начальным и конечным состояниями системы бесконечно мала.
- ↑ Простой называют закрытую термодеформационную систему, представляющую собой однородную изотропную среду (фазу) неизменного химического состава и массы, описываемую посредством переменных
- ↑ Сивухин Д. В., Термодинамика и молекулярная физика, 2005, с. 59.
- ↑ 1 2 Радушкевич Л. В., Курс термодинамики, 1971, с. 36.
- ↑ Базаров И. П., Термодинамика, 2010, с. 37.
- ↑ Условия интегрируемости дифференциальных полиномов подробно рассмотрены в книге Белоконь Н. И., Термодинамика, 1954, с. 137—138.
- ↑ Кричевский И. Р., Понятия и основы термодинамики, 1970, с. 270.
- ↑ Сычёв, 1991, с. 22.
- ↑ 1 2 Путилов К. А., Термодинамика, 1971, с. 13.
- ↑ Квасников, 2002, с. 43.
- ↑ 1 2 Сычёв, 1991, с. 24.
- ↑ Радушкевич Л. В., Курс термодинамики, 1971, с. 111.
- ↑ 1 2 Базаров И. П., Термодинамика, 2010, с. 27.
- ↑ Сычёв, 2009, с. 13.
- ↑ Кубо Р., Термодинамика, 1970, с. 21.
- ↑ Базаров И. П., Термодинамика, 2010, с. 27–29.
- ↑ Семенченко, 1966, с. 55.
- ↑ Сычёв, 2009, с. 14.
- ↑ 1 2 Кубо Р., Термодинамика, 1970, с. 20.
- ↑ Базаров И. П., Термодинамика, 2010, с. 38.
- ↑ Радушкевич Л. В., Курс термодинамики, 1971, с. 38.
- ↑ Глазов В. М., Основы физической химии, 1981, с. 29.
- ↑ Путилов К. А., Термодинамика, 1971, с. 40.
- ↑ Сейдж Б. Х., Термодинамика многокомпонентных систем, 1969, с. 54.
- ↑ Тамм М. Е., Третьяков Ю. Д., Физико-химические основы неорганической химии, 2004, с. 11.
- ↑ Lebon, 2008, p. 14.
- ↑ Жариков, 2005, уравнение (2.4)..
- ↑ Callen, 1985, p. 36.
- ↑ Сычёв, 2009, с. 257.
- ↑ Путилов К. А., Термодинамика, 1971, с. 125.
- ↑ Использование масс компонентов, а не масс составляющих систему веществ в качестве обобщённых координат в выражении для химической работы означает отказ от прямого рассмотрения влияния химических реакций на массы веществ, ибо химические превращения в системе уже учтены при подсчёте числа компонентов.
- ↑ Квасников И. А., Молекулярная физика, 2009, с. 31.
- ↑ Квасников, 2002, с. 22.
- ↑ 1 2 Петров Н., Бранков Й., Современные проблемы термодинамики, 1986, с. 66.
- ↑ 1 2 3 Tisza, 1966.
- ↑ Бэр Г. Д., Техническая термодинамика, 1977, с. 73.
- ↑ Залевски К., Феноменологическая и статистическая термодинамика, 1973, с. 10.
- ↑ Пригожин И., Кондепуди Д., Современная термодинамика, 2002, с. 52.
- ↑ Кубо Р., Термодинамика, 1970, с. 16.
- ↑ Жилин П. А., Рациональная механика сплошных сред, 2012.
- ↑ «…Понятия энергии, температуры, энтропии и химического потенциала вводятся одновременно и по отдельности определить их принципиально нельзя» (с. 48), «…Нельзя сначала определить внутреннюю энергию, а затем химический потенциал и энтропию. Все эти понятия могут быть введены только одновременно» (с. 140).
- ↑ 1 2 Петров Н., Бранков Й., Современные проблемы термодинамики, 1986, с. 43.
- ↑ Воронин Г. Ф., Основы термодинамики, 1987, с. 50.
- ↑ Callen, 1985, p. 28—29.
- ↑ 1 2 Мюнстер А., Химическая термодинамика, 1971, с. 69.
- ↑ Мюнстер А., Химическая термодинамика, 1971, с. 70.
- ↑ Мюнстер А., Химическая термодинамика, 1971, с. 67.
- ↑ Сам Гиббс эти постулаты в своей основной термодинамической работе «О равновесии гетерогенных веществ» формулировал по мере необходимости, как бы мимоходом, и не называл принимаемые им без доказательства утверждения ни аксиомами, ни постулатами.
- ↑ 1 2 Гиббс Дж. В., Термодинамика. Статистическая механика, 1982, с. 93.
- ↑ 1 2 3 Guggenheim, 1985, p. 15.
- ↑ 1 2 3 Callen, 1985, p. 35.
- ↑ Трусделл К., Термодинамика для начинающих, 1970.
- ↑ Трусделл К., Первоначальный курс рациональной механики сплошных сред, 1975.
- ↑ Truesdell, 1984, 1984.
- ↑ 1 2 Жилин П. А., Рациональная механика сплошных сред, 2012.
- ↑ Максимов, 2009.
- ↑ Максимов, 2009, с. 5.
- ↑ Максимов, 2009, с. 5—6.
- ↑ 1 2 3 Максимов, 2009, с. 7.
- ↑ 1 2 Петров Н., Бранков Й., Современные проблемы термодинамики, 1986, с. 71–72.
- ↑ Мюнстер А., Химическая термодинамика, 1971, с. 90–91.
- ↑ 1 2 Callen, 1985, p. 28.
- ↑ 1 2 3 Мюнстер А., Химическая термодинамика, 1971, с. 92.
- ↑ Сорокин В. С., Макроскопическая необратимость и энтропия. Введение в термодинамику, 2004, с. 55.
- ↑ Воронин Г. Ф., Основы термодинамики, 1987, с. 51.
- ↑ Петров Н., Бранков Й., Современные проблемы термодинамики, 1986, с. 67.
- ↑ Кубо Р., Термодинамика, 1970, с. 85—86.
- ↑ Buchdahl H. A., The Concepts of Classical Thermodynamics, 1966, p. 74.
- ↑ Зоммерфельд А., Термодинамика и статистическая физика, 1955, с. 52.
- ↑ В термодинамике различают аддитивность по размерам системы (длине упругого стержня или пружины, площади поверхности раздела, объёму) и аддитивность по массе. Ясно, что последнее понятие не универсально, и даже аддитивность экстенсивных переменных по объёму не гарантирует, что к этим переменным применимо представление об аддитивности по массе. Например, оно непригодно для аддитивных по объёму переменных фотонного газа — системы с нулевой массой.
- ↑ Петров Н., Бранков Й., Современные проблемы термодинамики, 1986, с. 62.
- ↑ Тер Хаар Д., Вергеланд Г., Элементарная термодинамика, 1968, с. 117.
- ↑ Ландау Л. Д., Лифшиц Е. М., Статистическая физика. Часть 1, 2002, с. 51.
- ↑ Falk, Jung, 1959, S. 156.
- ↑ Сычёв, 1991, с. 15.
- ↑ Воронин Г. Ф., Основы термодинамики, 1987, с. 53.
- ↑ Отрицательная температура. БСЭ, 3-е изд., 1975, т. 19.
- ↑ 1 2 3 4 Петров Н., Бранков Й., Современные проблемы термодинамики, 1986, с. 68.
- ↑ Callen, 1985, p. 30.
- ↑ Базаров И. П., Термодинамика, 2010, с. 92.
- ↑ Tisza, 1966, p. 125.
- ↑ 1 2 Новиков И. И., Термодинамика, 1984, с. 106.
- ↑ Базаров И. П., Термодинамика, 2010, с. 121.
- ↑ 1 2 Воронин Г. Ф., Основы термодинамики, 1987, с. 102.
- ↑ Мысленное допустимое (не противоречащее условиям существования системы) изменение энтропии, не зависящее от времени. Встречающееся в литературе определение вариации как отклонения от равновесия, допускаемого наложенными на систему связями (условиями), означает то же самое.
- ↑ Базаров И. П. и др., Термодинамика и статистическая физика, 1986, с. 26.
- ↑ Базаров И. П., Термодинамика, 2010, с. 29.
- ↑ 1 2 Хайтун С. Д., Кризис теории познания, 2014, с. 98.
- ↑ Хайтун С. Д., Кризис теории познания, 2014, с. 100.
- ↑ Хайтун С. Д., Кризис теории познания, 2014, с. 102.
- ↑ Lambert Frank L. A Brief Introduction to the Second Law and to Entropy for Chemistry Students
- ↑ Бриллюэн Л. Наука и теория информации. — М., 1960.
- ↑ Зарипов Р. Г., Новые меры и методы в теории информации, 2005, с. 175.
- ↑ 1 2 Кричевский И. Р., Петрянов И. В. Термодинамика для многих, 1975, с. 146.
- ↑ Гухман, 2010, с. 11.
- ↑ Ляшков В. И., Теоретические основы теплотехники, 2015, с. 10.
- ↑ Цирлин А. М., Методы оптимизации в необратимой термодинамике и микроэкономике, 2003, с. 19.
- ↑ Исаев С. И., Курс химической термодинамики, 1986, с. 18.
- ↑ Жуковский В. С., Термодинамика, 1983, с. 11.
- ↑ Леонова В. Ф., Термодинамика, 1968, с. 19—20.
- ↑ Трусделл К., Термодинамика для начинающих, 1970, с. 117.
- ↑ Свиридов В. В., Свиридов А. В., Физическая химия, 2016, с. 113.
- ↑ Максимов, 2009, с. 7.
- ↑ Максимов, 2009, с. 6.
- ↑ Игнатович В. Н., Введение в диалектико-материалистическое естествознание, 2007, с. 411.
- ↑ Гельфер Я. М., История и методология термодинамики и статистической физики, 1981, с. 228.
- ↑ Nernst Walther, Theoretische Chemie, 1900, S. 29.
- ↑ Кикоин И. К. Рассказы о физике и физиках, 1986, с. 33.
Литература[править | править код]
- Buchdahl H. A. The Concepts of Classical Thermodynamics. — Cambridge: Cambridge University Press, 1966. — XI + 223 p.
- Callen H. B. Thermodynamics and an Introduction to Thermostatistics. — 2nd ed. — N. Y. e. a.: John Wiley, 1985. — xvi + 493 p. — ISBN 0471862568, 9780471862567.
- Clausius R. Die mechanische Wärmetheorie. Band 1. — 3 Auflage. — Braunschweig: Druck und Verlag von Friedrich Vieweg und Sohn, 1887. — XVI + 403 p.
- Ehrenfest-Afanassjewa T. Zur Axiomatisierung des zweiten Hauptsatzes der Thermodynamik (нем.) // Zeitschrift für Physik. — 1925. — Vol. 33, Nr. 1. — P. 933–945.
- Ehrenfest-Afanassjewa T. Berichtigung zu der Arbeit: Zur Axiomatisierung des zweiten Hauptsatzes der Thermodynamik (нем.) // Zeitschrift für Physik. — 1925. — Vol. 34, Nr. 1. — P. 638.
- Ehrenfest-Afanassjewa T. Die Grundlagen der Thermodynamik. — Leiden: E.J. Brill, 1956. — XII + 131 с.
- Falk G., Jung H. Axiomatik der Thermodynamik (нем.) // Flügge S. (ed.). Encyclopedia of Physics / Flügge S. (Hrsg.). Handbuch der Physik. — Springer-Verlag, 1959. — Vol. III/2. Principles of Thermodynamics and Statistics / Band III/2. Prinzipien der Thermodynamik und Statistik, S. 119–175.
- Guggenheim E. A. Thermodynamics: An Advanced Treatment for Chemists and Physicists. — 7th ed. — Amsterdam: North-Holland, 1985. — xxiv + 390 p. — ISBN 0 444 86951 4.
- Lebon G., Jou D., Casas-Vázquez J. Understanding Non-equilibrium Thermodynamics: Foundations, Applications, Frontiers. — Berlin — Heidelberg: Springer, 2008. — xiii + 325 p. — ISBN 978-3-540-74251-7, 978-3-540-74252-4. — doi:10.1007/978-3-540-74252-4.
- Nernst Walther. Theoretische Chemie vom Standpunkte der avogadro’schen Regel und der Thermodynamik. — Dritte Auflage. — Stuttgart: Verlag von Ferdinand Enke, 1900. — xiv + 710 p.
- Noll W. The Foundations of Mechanics and Thermodynamics: Selected Papers. — Berlin — Heidelberg — New York: Springer-Verlag, 1974. — X + 324 p. — ISBN 978-3-642-65819-8.
- Reif F. Fundamentals of statistical and thermal physics. — McGraw-Hill, 1965.
- Tisza Laszlo. Generalized Thermodynamics. — Cambridge (Massachusetts) — London (England): The M.I.T. Press, 1966. — xi + 384 p.
- Truesdell C. The Tragicomical History of Thermodynamics, 1822–1854. — New York — Heidelberg — Berlin: Springer-Verlag, 1980. — xii + 372 p. — (Studies in the History of Mathematics and Physical Sciences. Vol. 4). — ISBN 978-1-4613-9446-4.
- Truesdell C., Bharatha S. The Concepts and Logic of Classical Thermodynamics as a Theory of Heat Engines. — New York — Heidelberg — Berlin: Springer-Verlag, 1977. — xvii + 154 p. — ISBN 3-540-07971-8.
- Truesdell C. Rational Thermodynamics. — New York—Berlin—Heidelberg—Tokyo: Springer-Verlag, 1984. — xviii + 578 p. — ISBN 0-387-90874-9.
- Wehrl Alfred. General properties of entropy (англ.) // Reviews of Modern Physics. — American Physical Society, 1978. — Vol. 50, no. 2. — P. 221–260. — doi:10.1103/RevModPhys.50.221.
- Алексеев Г. Н. Энергия и энтропия. — М.: Знание, 1978. — 192 с. — (Жизнь замечательных идей).
- Архаров А. М., Исаев С. И., Кожинов И. А. и др. Теплотехника / Под. общ. ред. В. И. Крутова. — М.: Машиностроение, 1986. — 432 с.
- Ауэрбах Ф. Царица мира и её тень. — 6-е изд. — Одесса: Матезис, 1913. — VIII + 50 с.
- Афанасьева-Эренфест Т. А. Необратимость, односторонность и второе начало термодинамики // Журнал прикладной физики. — 1928. — Vol. 5, № 3–4. — P. 3—30.
- Базаров И. П., Геворкян Э. В., Николаев П. Н. Термодинамика и статистическая физика. Теория равновесных систем. — М.: Издательство Московского университета, 1986. — 311 с.
- Базаров И. П. Термодинамика. — 5-е изд. — СПб.—М.—Краснодар: Лань, 2010. — 384 с. — (Учебники для вузов. Специальная литература). — ISBN 978-5-8114-1003-3.
- Белоконь Н. И. Термодинамика. — М.: Госэнергоиздат, 1954. — 416 с.
- Белоконь Н. И. Основные принципы термодинамики. — М.: Недра, 1968. — 112 с.
- Борн М. Критические замечания по поводу традиционного изложения термодинамики // Развитие современной физики. — М.: Наука, 1964. — С. 223—256.
- Бэр Г. Д. Техническая термодинамика. — М.: Мир, 1977. — 519 с.
- Волькенштейн М. В. Энтропия и информация. — М.: Наука, 1986. — 192 с. — (Проблемы науки и технического прогресса).
- Воронин Г. Ф. Основы термодинамики. — М.: Издательство Моск. университета, 1987. — 192 с.
- Гельфер Я. М. История и методология термодинамики и статистической физики. — 2-е изд., перераб. и доп. — М.: Высшая школа, 1981. — 536 с.
- Герасимов Я. И., Древинг В. П., Ерёмин Е. Н. и др. Курс физической химии / Под общ. ред. Я. И. Герасимова. — 2-е изд. — М.: Химия, 1970. — Т. I. — 592 с.
- Гиббс Дж. В. Термодинамические работы / Пер. с англ. под ред. проф. В. К. Семенченко. — М. — Л.: Гостехтеориздат, 1950. — 492 с. — (Классики естествознания).
- Гиббс Дж. В. Термодинамика. Статистическая механика / Отв. ред. Д. Н. Зубарев. — М.: Наука, 1982. — 584 с. — (Классики науки).
- Глазов В. М. Основы физической химии. — М.: Высшая школа, 1981. — 456 с.
- Гленсдорф П., Пригожин И. Термодинамическая теория структуры, устойчивости и флуктуаций. — М.: Мир, 1973. — 280 с.
- Гухман А. А. Об основаниях термодинамики. — 2-е изд., испр. — М.: Издательство ЛКИ, 2010. — 384 с. — ISBN 978-5-382-01105-9.
- Де Гроот С., Мазур П. Неравновесная термодинамика. — М.: Мир, 1964. — 456 с.
- Жариков В. А. Основы физической геохимии. — М.: Наука; Издательство МГУ, 2005. — 656 с. — (Классический университетский учебник). — ISBN 5-211-04849-0, 5-02-035302-7.
- Жилин П. А. Рациональная механика сплошных сред. — 2-е изд. — СПб.: Издательство Политехн. университета, 2012. — 584 с. — ISBN 978-5-7422-3248-3.
- Жуковский В. С. Термодинамика / Под ред. А. А. Гухмана. — М.: Энергоатомиздат, 1983. — 304 с.
- Залевски К. Феноменологическая и статистическая термодинамика: Краткий курс лекций / Пер. с польск. под. ред. Л. А. Серафимова. — М.: Мир, 1973. — 168 с.
- Зарипов Р. Г. Новые меры и методы в теории информации. — Казань: Издательство Казан. гос. техн. университета, 2005. — 364 с.
- Зоммерфельд А. Термодинамика и статистическая физика / Пер. с нем.. — М.: Издательство иностр. литературы, 1955. — 480 с.
- Игнатович В. Н. Введение в диалектико-материалистическое естествознание. — Киев: Экмо, 2007. — 468 с. — ISBN 978-966-8555-78-7.
- Исаев С. И. Курс химической термодинамики. — 2-е изд., перераб. и доп. — М.: Высшая школа, 1986. — 272 с.
- Квасников И. А. Молекулярная физика. — М.: Эдиториал УРСС, 2009. — 232 с. — ISBN 978-5-901006-37-2.
- Квасников И. А. Термодинамика и статистическая физика. Т. 1: Теория равновесных систем: Термодинамика. — 2-е изд., сущ. перераб. и доп. — М.: Едиториал УРСС, 2002. — 240 с. — ISBN 5-354-00077-7.
- Каратеодори К. Об основах термодинамики (1909) // Развитие современной физики — Отв. ред. Б. Г. Кузнецов. — М.: Наука, 1964. — 331 с. — С. 223—256.
- Кикоин И. К. Рассказы о физике и физиках. — Библиотечка «Квант». Выпуск 53. — М.: Наука, 1986. — 160 с.
- Клаузиус Р. Механическая теория тепла // Второе начало термодинамики. — М.—Л.: Гостехиздат, 1934. — С. 70—158.
- Кричевский И. Р. Понятия и основы термодинамики. — 2-е изд., пересмотр. и доп. — М.: Химия, 1970. — 440 с.
- Кричевский И. Р., Петрянов И. В. Термодинамика для многих. — М.: Педагогика, 1975. — 160 с. — (Библиотечка Детской энциклопедии «Учёные — школьнику»).
- Кубо Р. Термодинамика. — М.: Мир, 1970. — 304 с.
- Ландау Л. Д., Лифшиц Е. М. Статистическая физика. Часть 1. — 5-е изд. — М.: Физматлит, 2002. — 616 с. — (Теоретическая физика в 10 томах. Том 5). — ISBN 5-9221-0054-8.
- Леонова В. Ф. Термодинамика. — М.: Высшая школа, 1968. — 159 с.
- Ляшков В. И. Теоретические основы теплотехники. — М.: Курс; Инфра-М, 2015. — 328 с. — ISBN 978-5-905554-85-8, 978-5-16-0І0639-7.
- Максимов Л. А., Михеенков А. В., Полищук И. Я. Лекции по статистической физике. — Долгопрудный: МФТИ, 2009. — 224 с.
- Морачевский А. Г., Смирнова Н. А., Пиотровская Е. М. и др. Термодинамика равновесия жидкость—пар / Под ред. А. Г. Морачевского. — Л.: Химия, 1989. — 344 с. — ISBN 5-7245-0363-8.
- Мюнстер А. Химическая термодинамика / Пер. с нем. под. ред. чл.-корр. АН СССР Я. И. Герасимова. — М.: Мир, 1971. — 296 с.
- Новиков И. И. Термодинамика. — М.: Машиностроение, 1984. — 592 с.
- Осипов А.И., Уваров А.В. Энтропия и её роль в науке // Сетевой образовательный журнал. — 2004. — Т. 8, № 1. — С. 70—79.
- Петров Н., Бранков Й. Современные проблемы термодинамики. — Пер. с болг. — М.: Мир, 1986. — 287 с.
- Полянин А. Д., Полянин В. Д., Попов В. А. и др. Краткий справочник для инженеров и студентов. — М.: Международная программа образования, 1996. — 432 с. — ISBN 5-7753-0001-7.
- Пригожин И., Кондепуди Д. Современная термодинамика. От тепловых двигателей до диссипативных структур / Пер. с англ. — М.: Мир, 2002. — 462 с.
- Пригожин И., Стенгерс И. Порядок из хаоса: Новый диалог человека с природой. — М.: Прогресс, 1986. — 461 с.
- Путилов К. А. Термодинамика / Отв. ред. М. Х. Карапетьянц. — М.: Наука, 1971. — 376 с.
- Радушкевич Л. В. Курс термодинамики. — М.: Просвещение, 1971. — 288 с.
- Развитие современной физики. Сборник статей / Отв. ред. Кузнецов Б. Г.. — М.: Наука, 1964. — 331 с.
- Свиридов В. В., Свиридов А. В. Физическая химия. — СПб.: Лань, 2016. — 597 с. — ISBN 978-5-8114-2262-3.
- Свиридонов М. Н. Развитие понятия энтропии в работах Т. А. Афанасьевой-Эренфест // История и методология естественных наук. Выпуск X. Физика. — Издательство МГУ, 1971. — P. 112—129.
- Сейдж Б. Х. Термодинамика многокомпонентных систем. — М.: Недра, 1969. — 304 с.
- Семенченко В. К. [www.libgen.io/book/index.php?md5=FB60848D6D70A19DBF1154ECCAE49E7F Избранные главы теоретической физики]. — 2-е изд., испр. и доп. — М.: Просвещение, 1966. — 396 с. (недоступная ссылка)
- Сивухин Д. В. Общий курс физики. Т. II. Термодинамика и молекулярная физика. — 5-е изд., испр. — М.: ФИЗМАТЛИТ, 2005. — 544 с. — ISBN 5-9221-0601-5.
- Сорокин В. С. Макроскопическая необратимость и энтропия. Введение в термодинамику. — М.: ФИЗМАТЛИТ, 2004. — 174 с. — ISBN 5-9221-0507-8.
- Сычёв В. В. Дифференциальные уравнения термодинамики. — 2-е изд., перераб. — М.: Высшая школа, 1991. — 224 с. — ISBN 5-06-002071-1.
- Сычёв В. В. Дифференциальные уравнения термодинамики. — 3-е изд. — М.: Издательство МЭИ, 2010. — 256 с. — ISBN 978-5-383-00584-2.
- Сычёв В. В. Сложные термодинамические системы. — 5-е изд., перераб. и доп.. — М.: Издательский дом МЭИ, 2009. — 296 с. — ISBN 978-5-383-00418-0.
- Тамм М. Е., Третьяков Ю. Д. Неорганическая химия. Том 1. Физико-химические основы неорганической химии / Под. ред. акад. Ю. Д. Третьякова. — М.: Академия, 2004. — 240 с. — (Высшее профессиональное образование). — ISBN 5-7695-1446-9.
- Тер Хаар Д., Вергеланд Г. Элементарная термодинамика / Пер. с англ.. — М.: Мир, 1968. — 220 с..
- Термодинамика необратимых процессов. Лекции в летней международной школе физики им. Энрико Ферми / Под ред. Д. Н. Зубарева. — М.: Издательство иностранной литературы, 1962. — 427 с.
- Трайбус М. Термостатика и термодинамика. — М.: Энергия, 1971. — 503 с.
- Трусделл К. Термодинамика для начинающих // Механика. Периодический сборник переводов иностранных статей. — М.: Мир, 1970. — № 3 (121), с. 116—128.
- Трусделл К. Первоначальный курс рациональной механики сплошных сред / Пер. с англ. под. ред. П. А. Жилина и А. И. Лурье. — М.: Мир, 1975. — 592 с.
- Фен Дж. Машины, энергия, энтропия. — М.: Мир, 1986. — 335 с.
- Fermi, E., Thermodynamics, Prentice Hall (1937). — Русский перевод: Ферми, Энрико, Термодинамика, Харьков: Издательство Харьковского университета, 1969. — 140 с.
- Физическая энциклопедия / Гл. ред. А. М. Прохоров. — М.: Большая Российская энциклопедия, 1998. — Т. 5. — 760 с. — ISBN 5-85270-101-7.
- Хаазе Р. Термодинамика необратимых процессов. — М.: Мир, 1967. — 544 с.
- Черноуцан А. И. Краткий курс физики. — М.: ФИЗМАТЛИТ, 2002. — 320 с. — ISBN 5-9921-0292-3.
- Хазен А. М. Разум природы и разум человека. — М.: РИО «Мособлполиграфиздата»; НТЦ «Университетский», 2000. — 600 с. — ISBN 5-7953-0044-6.
- Хайтун С. Д. [www.libgen.io/book/index.php?md5=BB7A4DF3D04E410AB163CF2D15B4501C Кризис науки как зеркальное отражение кризиса теории познания: Кризис теории познания]. — М.: Ленанд, 2014. — 448 с. — ISBN 978-5-9710-1296-2. (недоступная ссылка)
- Цирлин А. М. Методы оптимизации в необратимой термодинамике и микроэкономике. — М.: Физматлит, 2003. — 416 с.
- Шамбадаль П. Развитие и приложение понятия энтропии / Пер. с франц. — М.: Наука, 1967. — 279 с.
Ссылки[править | править код]
- Термодинамическая энтропия
- Статья «Эволюция как сопротивление энтропии»
|
|
Некоторые внешние ссылки в этой статье ведут на сайты, занесённые в спам-лист. Эти сайты могут нарушать авторские права, быть признаны неавторитетными источниками или по другим причинам быть запрещены в Википедии. Редакторам следует заменить такие ссылки ссылками на соответствующие правилам сайты или библиографическими ссылками на печатные источники либо удалить их (возможно, вместе с подтверждаемым ими содержимым). Список проблемных ссылок
|
[Править] Построение графика изменения энтропии
Основная статья: адиабатический
процесс
Следующее
уравнение может быть использовано для
построения графика изменения энтропии
на диаграмме P—V:
![]()
Здесь
два замечания:
-
это не определение энтропии (но выведено
из него); -
предполагается, что CV и
CP постоянные, что на самом
деле не так.
4.5. Энтропия
Изменение
энтальпии системы не может служить
единственным критерием самопроизвольного
осуществления химической реакции,
поскольку многие эндотермические
процессы протекают самопроизвольно.
Иллюстрацией этого служит растворение
некоторых солей (например, NH4NO3)
в воде, сопровождающееся заметным
охлаждением раствора. Необходимо
учитывать еще один фактор, определяющий
способность самопроизвольно переходить
из более упорядоченного к менее
упорядоченному (более хаотичному)
состоянию.
Энтропия
(S) – термодинамическая функция состояния,
которая служит мерой беспорядка
(неупорядоченности) системы. Возможность
протекания эндотермических процессов
обусловлена изменением энтропии, ибо
в изолированных системах энтропия
самопроизвольно протекающего процесса
увеличивается ΔS > 0 (второй закон
термодинамики).
![]()
|
|
|
Модель 4.8. |

Л.
Больцман
определил энтропию как термодинамическую
вероятность состояния (беспорядок)
системы W. Энтропия связана с
термодинамической вероятностью
соотношением:
|
Размерность
энтропии 1 моля вещества совпадает с
размерностью газовой постоянной R и
равна Дж∙моль–1∙K–1.
Изменение энтропии *)
в необратимых и обратимых процессах
передается соотношениями ΔS > Q / T
и ΔS = Q / T. Например, изменение
энтропии плавления равно теплоте
(энтальпии) плавления ΔSпл = ΔHпл/Tпл.
Для химической реакции изменение
энтропии аналогично изменению энтальпии
|
|
*)
термин энтропия был введен Клаузиусом
(1865 г.) через отношение Q / T (приведенное
тепло).
Здесь
ΔS°
соответствует энтропии стандартного
состояния. Стандартные энтропии простых
веществ не равны нулю. В отличие от
других термодинамических функций
энтропия идеально кристаллического
тела при абсолютном нуле равна нулю
(постулат Планка),
поскольку W = 1.
Энтропия
вещества или системы тел при определенной
температуре является абсолютной
величиной. В табл. 4.1 приведены стандартные
энтропии S°
некоторых веществ.
|
Соединение
|
||||||||||||||||||||||||||||||||||||||||||||
|
Таблица
Стандартные |
Из
табл. 4.1 следует, что энтропия зависит
от:
-
агрегатного
состояния вещества. Энтропия увеличивается
при переходе от твердого к жидкому и
особенно к газообразному состоянию
(вода, лед, пар). -
изотопного
состава (H2O
и D2O). -
молекулярной
массы однотипных соединений (CH4,
C2H6,
н-C4H10). -
строения
молекулы (н-C4H10,
изо-C4H10). -
кристаллической
структуры (аллотропии) – алмаз, графит.
Наконец,
рис. 4.3 иллюстрирует зависимость энтропии
от температуры.
|
|
|
Рисунок Зависимость |

Следовательно,
стремление системы к беспорядку
проявляется тем больше, чем выше
температура. Произведение изменения
энтропии системы на температуру T ΔS
количественно оценивает эту тендецию
и называется энтропийным фактором.
![]()
|
|
|
Модель 4.9. |

Энтропия.
Изменение энтропии в химических и
фазовых переходах.
Состояние любой системы может быть
охарактеризовано значениями непосредственно
измеряемых параметров (р, Т и др.). Это
характеристика макросостояния
системы. Состояние системы
может быть описано также
характеристиками каждой частицы системы
(атома, молекулы): координаты, частота
колебания, частота вращения и т.д. Это
характеристика микросостояния
системы. Системы состоят из
очень большого числа частиц, поэтому
одному макросостоянию будет отвечать
огромное число различных микросостояний.
Это число называется термодинамической
вероятностью состояния и обозначается
(W).
Термодинамическая вероятность связана
с другим свойством вещества – энтропией
(S) – формулой Больцмана
 (2.13)
(2.13)
где R –
универсальная газовая постоянная, а NA
– постоянная Авогадро. Измеряется
энтропия в Дж/(моль.К).
Физический смысл: энтропия является
мерой неупорядоченности состояния
системы. Энтропия системы
увеличивается во всех процессах, когда
возрастает неупорядоченность (нагревание,
растворение, испарение, реакции разложения
и т.п.) и уменьшается в процессах, идущих
с увеличением упорядоченности (охлаждение,
кристаллизация, сжатие и т.п.).
Энтропия является функцией состояния,
но в отличие от большинства других
термодинамических функций возможно
экспериментальное определение абсолютного
значения энтропии вещества. Эта
возможность основана на постулате
М.Планка, согласно которому «при
абсолютном нуле энтропия идеального
кристалла равна нулю» (третий закон
термодинамики).
Температурная
зависимость энтропии вещества представлена
качественно на рис.2.1.

На рис.2.1 видно, что при нуле
Кельвина энтропия вещества равна нулю.
При повышении температуры энтропия
плавно увеличивается, а в точках фазовых
переходов имеет место скачкообразное
увеличение энтропии, определяемое
соотношением
![]()
(2.14)
где Δ ф..п.S, Δф.п.Н
и Тф.п. изменение энтропии,
энтальпии и температура фазового
перехода, соответственно.
Энтропию вещества B в стандартном
состоянии обозначают ![]() .
.
Для многих веществ абсолютные значения
стандартных энтропий определены и
приводятся в справочных изданиях.
Энтропия, также как внутренняя энергия
и энтальпия, является функцией состояния,
поэтому изменение энтропии системы в
процессе не зависит от его пути и
определяется только начальным и конечным
состоянием системы. Изменение энтропии
в ходе химической реакции (2.10) может
быть найдено, как разность суммы энтропий
продуктов реакции и суммы энтропий
исходных веществ
![]()
Понятие энтропии используется в одной
из формулировок второго закона
термодинамики: в изолированных
системах могут самопроизвольно протекать
только процессы, идущие с увеличением
энтропии (ΔS>0). Под изолированными
системами понимаются системы, не
обменивающиеся с окружающей средой ни
веществом, ни энергией. Системы, в которых
протекают химические процессы, к
изолированным системам не относятся,
т.к. они обмениваются с окружающей средой
энергией (тепловой эффект реакции).
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
В качестве другого примера графического дифференцирования укажем на вычисление температурного коэффициента электродвижущей силы гальванического элемента (для определения теплового эффекта или изменения энтропии в токообразующем процессе). [c.446]
| Рис. П1,3. Графическое вычисление энтропии этилена. | 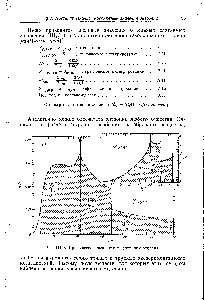 |
На рис. Ill, 3, иллюстрирующем графический метод расчета энтропии на примере вычисления энтропии этилена, изображены кривые Ср/Т = ф(Т) для твердого, жидкого и газообразного этилена штриховкой показаны площади, соответствующие приростам энтропии при нагревании . (Изменения энтропии при агрегатных превращениях, естественно, не могут получить отражения на данной диаграмме.) [c.99]
Второй и третий интегралы, которые соответственно равны изменению энтропии при нагревании двух модификаций твердого хлористого водорода, определяем графическим интегрированием. Значения второго и третьего интегралов соответственно равны 29,54 и 21,17 Дж/(моль К)- Четвертый и пятый интегралы могут быть вычислены как графическим интегрированием, так и аналитическим путем. Воспользуемся для их вычисления аналитическим методом. Зависимость теплоемкости жидкого хлористого водорода от температуры можно выразить уравнением [c.235]
Примерами графического интегрирования служат вычисление энтальпии и энтропии вещества по температурной завнсимости теплоемкости первой — по графику С,, = / (Г), второй — по графику Ср/Т =- / (Т) нли С,, = / (1п Т). На рис. 201 приведен пример графического интегрирования для определения теплового эффекта химического процесса. [c.448]
Величины остаточных свойств можно получить непосредственно из экспериментальных данных. Так, калориметрические измерения энтальпии и теплоемкости дают собственно остаточную энтальпию Д// и остаточную энтропию Д5. Однако количество подобных данных, особенно для смесей, ограничено. Некоторые примеры экстремального воздействия давления на теплоемкость показаны на рис. 1.3 и 1.4. На рис. 11.2 в графической форме представлен ряд измеренных величин остаточных свойств этилена. В примере 11.4 экспериментальные данные о зависимости PVT для азота применены для оценки остаточных энтальпий и энтропий результаты этих вычислений сопоставляются с результатами расчетов, выполненных на основе некоторых уравнений состояния. [c.512]
Находя температуру, при которой давление пара твердой и жидкой фаз одинаково, мы определим точку плавления. Энтропия плавления может быть определена обычным способом как разность энтропии твердого тела и жидкости при температуре плавления. Изображая графически максимальную работу как функцию объема и находя две точки, в которых кривая имеет общую касательную, найдем объемы сосуществующих фаз (жидкость —пар), а также и соответствующие упругости паров. Однако на практике вычисления упрощаются тем, что для решения вопроса привлекают уравнение Ван-дер-Ваальса. [c.618]
После того как вычислена теплота адсорбции по (XI.15) или теплота растворения по (XI.15а), можно приступить к вычислению энтропии адсорбции Д5а или энтропии растворения Д5а, исходя из значений В, т. е. отрезка, отсекаемого прямой графика функции Ig (Г, К) =/(1/7 ) на оси ординат [рис. XI.3 формула (XI.13)]. В этом случае AS—2,3 RB. Однако такой способ расчета энтропии ненадежен, поскольку ненадежно графическое определение константы В. [c.256]
Третий закон используется при вычислениях абсолютных энтропий простых и сложных веществ. Абсолютной энтропией называется энтропия, отсчитанная относительно = 0. Для вычисления необходимо графическое или аналитическое интегрирование уравнения (VI.4). Однако нижним пределом интегрирования является = О К, поэтому [c.97]
На практике вычисление энтропии часто проводят графическим спосо- [c.188]
На рис. 31 представлена зависимость величин Гд5г, вычисленных графически, от концентрации толуола, т. е. от степени разбавления. Мы видим, что парциальная молярная энтропия каучука непрерывно возрастает с разбавлением. Полученные значения также значительно больше идеальной энтропии смешения, вычисленной по формуле = = — 1п N2. [c.117]
Освоив технику графического интегрирования, перейдем к основному интересующему нас вопросу — вычислению энтропии из [c.164]
Изменение энтропии в процессе, проходящем не при постоянной температуре, а при непрерывно изменяющейся, как это имеет место при нагревании вещества, можно вычислить тем же способом графического интегрирования. Площадь под кривой теплоемкости делится на элементарные площади и для каждой подсчитывается изменение энтальпии. Разделив такое элементарное изменение энтальпии на температуру, среднюю между температурами начала и конца элементарного интервала температур, получим изменение энтропии при нагревании вещества в элементарном интервале температур АТ. Просуммировав все частные от деления, получим изменение энтропии в интересующем нас интервале температуры. Проще можно поступить следующим образом разделить все теплоемкости на соответствующие им температуры и далее графическое интегрирование (в данном случае уже числовое интегрирование) проводить точно так же, как и с изменениями энтальпии. Практически для вычисления изменения энтропии при нагревании вещества от одной температуры до другой, если известен ход теплоемкости, следует поступить так разбить весь интервал на желаемое число частей по АТ градусов (5—10°), определить по графику темплоемкости, отвечающие каждой температуре, значения теплоемкости разделить на соответствующие им температуры, просуммировать частные от деления, за исключением первой и последней, сумму умножить на число градусов в АТ, к этой сумме прибавить полусумму первого и последних частных, умноженную на АТ. Если интегрирование начинается с 0°К, то на АТ умножается только половина последнего частного от деления. [c.165]
В качестве нижнего предела в этих уравнениях можно взять любую температуру. В большинстве случаев расчет ведут относительно Т° = 273,2, причем значения энтропии и энтальпии обычно представляют в графической форме, в виде диаграмм Н — 5 и Т — S. Эти диаграммы широко применяются для технологических расчетов, так как многие вычисления могут быть сведены к простому отсчету по диаграмме. [c.164]
Примерами графического интегрирования служат вычисление энтальпии и энтропии вещества по температурной зависимости теплоемкости первой — по графику Ср =/ (Г), второй — по графику [c.19]
Таким образом, измеряя теплоемкости элементов и чистых соединений от комнатной температуры до возможно более низкой температуры, например до точки плавления водорода (15° К) или до точки кипения кислорода (90° К), и экстраполируя полученные величины до абсолютного нуля, возможно составить таблицу стандартных энтропий всех известных чистых веществ, которую можно затем использовать для вычисления AS любой интересующей нас xiimh-ческой реакции. Для любого процесса при заданной температуре величина AS получается вычитанием суммы энтропий веществ, составляющих начальное состояние системы, из суммы энтропий веществ в конечном состоянии. Так как при низких температурах теплоемкости очень быстро убывают и стремятся к нулю с приближением температуры к абсолютному нулю, то необходимо прибегать к графическому интегрированию уравнения (6.5). Методы этого интегрирования, описанные в деталях у Льюиса и Рендалла и у Паркса и Хаффмана [1], рассматриваются в гл. VII в связи с применением закона теплоемкостей Дебая для экстраполяции до 0° К. [c.83]
Робинсон и Блисс [198] дали законченное аналитическое выражение для вычисления изотермического изменения U, Н и S по трем уравнениям состояния, определяющим р, а именно—по уравнениям Ван-дер-Ваальса, Воля и Битти — Бриджмена. Сравнение вычисленных данных для трех различных газов со значениями, найденными из данных p v—7 графическим методом, показывает, что уравнение Битти — Бриджмена является наилучшим по сравнению с двумя остальными, в особенности для вычисления энтальпии. Но даже с этим уравнением отклонение все же составляло 7 /д для энтропии и 12 / (а в некоторых случаях и более) — для энтальпии. [c.272]
Изменения энтропии и изобарно-изотермического потенциала при растворении КС1 в смесях НгО—СН3ОН находились по методу [2]. Коэффициенты активности хлористого калия в смешанных растворителях вода—метиловый спирт, необходимые для вычисления АСраств, заимствованы из работы Латимера и Сланского [3]. Графические зависимости всех трех величин от моляльной концентрации электролита т для пяти составов смесей вода — метанол изображены на рис. 1. Кривая 1 для АЯраств КС1 В воде построена по данным [4]. [c.111]
Поскольку давление паров не было измерено с достаточной точностью, коэффициенты активности ух и Уг находили из уравнений (27а) и (276), а среднюю молярную избыточную свободную энтальпию 0 вначале определяли из уравнения (22а). После этого среднюю молярную избыточную энтальпию рассчитывали по уравнению (29а). Величину устанавливали графически по тангенсу угла наклона прямой, изображающей зависимость величины функции (0 /ЯТ)х от температуры (рис. 66). Наконец, среднюю молярную избыточную энтроп ю5 находили из уравнения (30). Результаты этих вычислений представлены на рис. 67. [c.135]
Температуры переходов определены графически (рис. 3). Энтальпия перехода kIV—>-кП1 измерена методом непрерывного ввода энергии [7] в трех опытах (табл. 3). В табл. 2 приведен средний результат и указано среднеарифметическое отклонение. Энтропия перехода вычислена по значениям энтальпии и температуры его. Оценка энтальпий переходов кП1—>-кП и кП—>-к1 выполнена путем графического интегрирования С°р по температуре в интервале указанных переходов по экспериментально полученным кривым теплоемкости и нормальным кривым °p = f (T), показанным на рис. 3 пунктиром, причем AIi°ir получалась как разность энтальпий, вычисленных по этим кривым. Энтропии переходов оценивали аналогично, но интегрировали С р = = /(1пГ). Полученные значения AH°tr и AS°tr приведены вместе с гра-( )нческой ошибкой интегрирования. Следует отметить, что энтальпии и энтропии рассмотренных переходов малы. Полученные данные не позволяют сделать однозначных выводов относительно природы переходов. Однако можно высказать предположение, что все они относятся к переходам типа порядок5= беспорядок и обусловлены изменением взаимной ориентации молекул в кристаллической решетке. В известной мере аналогичную картину переходов получили Чанг и Веструм [9] при калориметрическом изучении С°р тиомочевины. Используя полученные результаты и данные кристаллографических исследований [10], они отнесли наблюдаемые превращения к переходам типа порядок гй беспо-рядок, объяснив их появление небольшими различными поворотами молекул в кристалле. [c.25]
Курс физической химии Том 1 Издание 2 (1969) — [
c.95
]
Курс физической химии Том 1 Издание 2 (копия) (1970) — [
c.95
]
| Термодинамические величины |
|---|

|
| Статья является частью серии «Термодинамика». |
| Энтропия |
| Количество теплоты |
| Термодинамическая работа |
| Химический потенциал |
| См. также: Термодинамические потенциалы. |
| Разделы термодинамики |
| Начала термодинамики |
| Уравнение состояния |
| Термодинамические величины |
| Термодинамические потенциалы |
| Термодинамические циклы |
| Фазовые переходы |
| править |
Термодинами́ческая энтропи́я S, часто просто именуемая энтропия, в химии и термодинамике является функцией состояния термодинамической системы; её существование постулируется вторым началом термодинамики.
Термодинамическое определение энтропии
Понятие энтропии было впервые введено в 1865 году Рудольфом Клаузиусом. Он определил изменение энтропии термодинамической системы при обратимом процессе как отношение изменения общего количества тепла ΔQ к величине абсолютной температуры T:

Рудольф Клаузиус дал величине S имя «энтропия», происходящее от греческого слова τρoπή, «изменение» (изменение, превращение, преобразование). Данное равенство относится к изменению энтропии, не определяя полностью саму энтропию.
Эта формула применима только для изотермического процесса (происходящего при постоянной температуре). Её обобщение на случай произвольного квазистатического процесса выглядит так:
,
где 
Необходимо обратить внимание на то, что рассматриваемое термодинамическое определение применимо только к квазистатическим процессам (состоящим из непрерывно следующих друг за другом состояний равновесия).
Поскольку энтропия является функцией состояния, в левой части равенства стоит её полный дифференциал. Напротив, количество теплоты является функцией процесса, в котором эта теплота была передана, поэтому
Энтропия, таким образом, согласно вышеописанному, определена вплоть до произвольной аддитивной постоянной. Третье начало термодинамики позволяет определить её точнее: предел величины энтропии равновесной системы при стремлении температуры к абсолютному нулю полагают равным нулю.
Статистическое определение энтропии: принцип Больцмана
В 1877 году Людвиг Больцман нашёл, что энтропия системы может относиться к количеству возможных «микросостояний» (микроскопических состояний), согласующихся с их термодинамическими свойствами. Рассмотрим, например, идеальный газ в сосуде. Микросостояние определено как позиции и импульсы (моменты движения) каждого составляющего систему атома. Связность предъявляет к нам требования рассматривать только те микросостояния, для которых:
(I) месторасположения всех частей расположены в рамках сосуда,
(II) для получения общей энергии газа кинетические энергии атомов суммируются.
Больцман постулировал, что:

где константу k=1,38•10–23 Дж/К мы знаем теперь как постоянную Больцмана, а Ω является числом микросостояний, которые возможны в имеющемся макроскопическом состоянии (статистический вес состояния). Этот постулат, известный как принцип Больцмана, может быть оценен как начало статистической механики, которая описывает термодинамические системы, используя статистическое поведение составляющих их компонентов. Принцип Больцмана связывает микроскопические свойства системы (Ω) с одним из её термодинамических свойств (S).
Согласно определению Больцмана, энтропия является просто функцией состояния. Так как Ω может быть только натуральным числом (1,2,3,…), то энтропия Больцмана должна быть положительной — исходя из свойств логарифма.
Понимание энтропии как меры беспорядка
Существует мнение, что мы можем смотреть на Ω и как на меру беспорядка в системе. В определённом смысле это может быть оправдано, потому что мы думаем об «упорядоченных» системах как о системах, имеющих очень малую возможность конфигурирования, а о «беспорядочных» системах, как об имеющих очень много возможных состояний. Собственно, это просто переформулированное определение энтропии как числа микросостояний на данное макросостояние.
Рассмотрим, например, распределение молекул идеального газа. В случае идеального газа наиболее вероятным состоянием, соответствующим максимуму энтропии, будет равномерное распределение молекул. При этом реализуется и максимальный “беспорядок”, т.к. при этом будут максимальные возможности конфигурирования.
Границы применимости понимания энтропии как меры беспорядка
Подобное определение беспорядка термодинамической системы как количества возможностей конфигурирования системы фактически дословно соответствует определению энтропии как числа микросостояний на данное макросостояние. Проблемы начинаются в двух случаях:
- когда начинают смешивать различные понимания беспорядка, и энтропия становится мерой беспорядка вообще;
- когда понятие энтропии применяется для систем, не являющихся термодинамическими.
В обоих этих случаях применение понятия термодинамической энтропии совершенно неправомерно[1].
Рассмотрим оба пункта более подробно.
Рассмотрим пример термодинамической системы – распределение молекул в поле тяготения. В этом случае наиболее вероятным распределением молекул будет распределение согласно барометрической формуле Больцмана. Другой пример – учёт электромагнитных сил взаимодействия между ионами. В этом случае наиболее вероятным состоянием, соответствующим максимуму энтропии, будет упорядоченное кристаллическое состояние, а совсем не “хаос”. (Термин “хаос” здесь понимается в смысле беспорядка – в наивном смысле. К хаосу в математическом смысле как сильно неустойчивой нелинейной системе это не имеет отношения, конечно.)
Рассмотрим случай с кристаллической решёткой более подробно. Кристаллическая решётка может быть и в равновесном, и в неравновесном состоянии, как и любая термодинамическая система. Скажем, возьмём следующую модель – совокупность взаимодействующих осцилляторов. Рассмотрим некоторое неравновесное состояние: все осцилляторы имеют
одинаковое отклонение от положения равновесия. С течением времени эта система перейдёт в состояние ТД равновесия, в котором отклонения (в каждый момент времени) будут подчинены некоторому распределению типа Максвелла (только это распределение будет для отклонений, и оно будет зависеть от типа взаимодействия осцилляторов). В таком случае максимум энтропии будет действительно реализовывать максимум возможностей конфигурирования, т.е. – беспорядок согласно вышеуказанному определению. Но данный “беспорядок” вовсе не соответствует “беспорядку” в каком-либо другом понимании, например, информационному. Такая же ситуация возникает и в примере с кристаллизацией переохлаждённой жидкости, в которой образование структур из “хаотичной” жидкости идёт параллельно с увеличением энтропии.
Это неверное понимание энтропии появилось во время развития теории информации, в связи с парадоксом термодинамики, связанным с мысленным экспериментом т.н. “демона Максвелла”. Суть парадокса заключалась в том, что рассматривалось два сосуда с разными температурами, соединённых узкой трубкой с затворками, которыми управлял т.н. “демон”. “Демон” мог измерять скорость отдельных летящих молекул, и т.о. избирательно пропускать более быстрые в сосуд с высокой температурой, а более медленные – в сосуд с низкой. Из этого мысленного эксперимента вытекало кажущееся противоречие со вторым началом термодинамики.
Парадокс может быть разрешён при помощи теории информации. Для измерения скорости молекулы “демон” должен был бы получить информацию о её скорости. Но всякое получение информации – материальный процесс, сопровождающийся возрастанием энтропии. Количественный анализ, проведённый, например, в [2] показал, что приращение энтропии при измерении превосходит по абсолютной величине уменьшение энтропии, вызванное перераспрелением молекул “демоном”.
Однако многие учёные стали отождествлять информацию с “отрицательной энтропией” ввиду совпадения выражений для этих понятий. Начало этому заблуждению положил Н. Винер [3] (стр. 23). На самом деле энтропия не является мерой дезорганизации, мерой беспорядка и хаоса, а информация совсем не является мерой упорядоченности, организованности, порядка.
Рассмотрим второй случай неверного применения понятия энтропии.
Рассмотрим, например, набор 10 монет, каждая из которых может находиться либо в состоянии «орёл», либо в состоянии «решка». Наиболее «упорядоченным» макроскопическим состоянием будет являться или 10 «орлов», или 10 «решек»; для каждого результата в каждом случае имеется только одна возможная конфигурация. И наоборот, наиболее «неупорядоченное» состояние содержит 5 «орлов» и 5 «решек», и здесь 10C5 = 252 способов для получения этого результата (см. комбинаторика.)
Безусловно, этот пример также некорректен, т.к. система монет не является термодинамической системой, и поэтому термодинамическая энтропия системы в обоих случаях (как бы ни были перевёрнуты монеты) окажется, конечно, одинаковой. Т.о., разбросанные по комнате стулья не имеют отношения к термодинамической энтропии, хотя и могут иметь отношение к энтропии информационной.
Впрочем, это легко подтвердить на практике: замкнутая система 10 монет, перевёрнутых орлами вверх, самопроизвольно не перейдёт в систему хаотично перевёрнутых монет.
Энтропия в открытых системах
В силу второго начала термодинамики, энтропия 






В стационарных системах обычно 


Суммарное изменение энтропии открытой системы будет равно:

Если всё время 

Измерение энтропии
В реальных экспериментах очень трудно измерить энтропию системы. Техники измерения базируются на термодинамическом определении энтропии и требуют экстремально аккуратной калориметрии.
Для упрощения мы будем исследовать механическую систему, термодинамические состояния которой будут определены через её объем V и давление P. Для измерения энтропии определенного состояния мы должны сперва измерить теплоёмкость при постоянных объёме и давлении (обозначенную CV и CP соответственно), для успешного набора состояний между первоначальным состоянием и требуемым. Тепловые ёмкости связаны с энтропией S и с температурой T согласно формуле:

где нижний индекс X относится к постоянным объёму и давлению. Мы можем проинтегрировать для получения изменения энтропии:

Таким образом, мы можем получить значение энтропии любого состояния (P,V) по отношению к первоначальному состоянию (P0,V0). Точная формула зависит от нашего выбора промежуточных состояний. Для примера, если первоначальное состояние имеет такое же давление, как и конечное состояние, то

В добавление, если путь между первым и последним состояниями лежит сквозь любой фазовый переход первого рода, скрытая теплота, ассоциированная с переходом, должна также учитываться.
Энтропия первоначального состояния должна быть определена независимо. В идеальном варианте выбирается первоначальное состояние как состояние при экстремально высокой температуре, при которой система существует в виде газа. Энтропия в этом состоянии подобна энтропии классического идеального газа плюс взнос от молекулярных вращений и колебаний, которые могут быть определены спектроскопически.
Построение графика изменения энтропии
Основная статья: адиабатический процесс
Следующее уравнение может быть использовано для построения графика изменения энтропии на диаграмме P-V:

Здесь два замечания: (1) это не определение энтропии (но выведено из него), (2) предполагается, что CV и CP постоянные, что на самом деле не так.
См. также
- Термодинамика чёрных дыр
- Тепловая смерть
- Демон Максвелла
- Термодинамические потенциалы
- Стрела времени
- Информационная энтропия
- Негэнтропия
- Энергетическое определение энтропии
- Список статей по энтропии
Литература
- Fermi, E., Thermodynamics, Prentice Hall (1937)
- Reif, F., Fundamentals of statistical and thermal physics, McGraw-Hill (1965)
- Шамбадаль П. Развитие и приложение понятия энтропии. – М.: Наука, 1967. – 280 с.
- Волькенштейн М.В. Энтропия и информация. – М.: Наука, 1986. – 192 с.
- ↑ http://www.entropysite.com/
- ↑ Бриллюэн Л. Наука и теория информации. – М., 1960.
- ↑ Винер Н. Кибернетика. – М.: Советское радио, 1958.
Ссылки
- Термодинамическая энтропия
- Entropy and chaos (англ.)
- Роль Термодинамики в современном мире. Второе начало термодинамики
- Статья “Эволюция как сопротивление энтропии”
|
Выделить Энтропия (в термодинамике) и найти в:
|
|
|
- Страница 0 – краткая статья
- Страница 1 – энциклопедическая статья
- Разное – на страницах: 2 , 3 , 4 , 5
- Прошу вносить вашу информацию в «Энтропия (в термодинамике) 1», чтобы сохранить ее
