
Андрей Геннадьевич Блохин
Эксперт по предмету «Физика»
Задать вопрос автору статьи
Энтропия – это фундаментальная физическая величина. Введение этого понятия завершило этап становления понятийного аппарата термодинамики. Следующим этапом развития этой науки было выяснение физического смысла энтропии.
Установление принципа Больцмана (формулы Больцмана) и таким образом связи между термодинамикой и статистической физикой, позволило энтропии покинуть пределы физики и войти в другие области знаний.
Энтропия – это общезначимое понятие, применяемое во множестве наук, например, в:
- физике;
- химии;
- билогии;
- космологии;
- теории информации.
Введено это понятие было Р. Клаузиусом. Важные работы посвятил энтропии Л. Больцман, М. Планк.
Определение 1
Функция состояния полным дифференциалом которой является δQ/T называется энтропией ($S$):
$dS=frac{delta Q}{T}left( 1 right)$,
где $delta Q$ – элементарное тело, получаемое термодинамической системой; $T$- термодинамическая температура.
Замечание 1
Отметим, что формула (1) справедлива только для обратимых процессов. Например, для процессов, проводимых в идеальном газе.
Важно, что выражение (1) определяет не саму энтропию, а ее изменение, или разность энтропий. При помощи данной формулы можно вычислить, каково изменение энтропии, если термодинамическая система переходит из одного состояния в другое, но нельзя понять, какова энтропия каждого из этих состояний.
Напомним формулу Больцмана для энтропии, так как она нам поможет понять физический смысл, процессов, которые происходят с энтропией:
$S=klnW, left( 2 right)$
где ($W$) – термодинамическая вероятность (статистический вес состояния), то есть количество способов, реализации данного состояния термодинамической системы. $W$ ≥1; $k$- постоянная Больцмана.
Изменение энтропии, исходя из статистической формулы Больцмана, найдем как:
$S_{2}-S_{1}=klnfrac{W_{2}}{W_{1}}left( 2.1 right)$).
«Изменение энтропии» 👇
Вычисление изменения энтропии в изотермическом процессе
Вычислим изменение энтропии в одном моле идеального газа при изотермическом процессе ($T=const$). Оттолкнемся от первого начала термодинамики в дифференциальной форме:
$delta Q=dU+pdV, left( 3 right)$,
где $dU$ – изменение внутренней энергии идеального газа; $pdV$=$delta A$ – работа, совершаемая газом.
Найдем отношение всех слагаемых выражения (3) и температуры:
$frac{delta Q}{T}=C_{V}frac{dT}{T}+frac{p}{T}dVleft( 4 right)$.
где $C_V$ – теплоемкость газа при $V=const$.
Для идеального газа справедливо уравнение Менделеева – Клапейрона, учитывая, что у нас 1 моль газа:
$pV=RTto frac{p}{T}=frac{R}{V}left( 5 right)$.
Учтем:
$frac{dT}{T}=dleft( ln T right),, frac{dV}{V}=dleft( ln V right)left( 6 right)$.
в этом случае мы можем записать для уравнения (4):
$frac{delta Q}{T}=dleft( C_{V}lnT+RlnV right)left( 7 right)$.
По определению (1), и принимая во внимание (7), запишем:
$dS=dleft( C_{V}lnT+RlnV right)left( 8 right)$.
Формула (8) показывает нам, что в изотермическом процессе изменяется только второе слагаемое правой части:
- при увеличении объема энтропия увеличивается,
- с уменьшением объема энтропия уменьшается.
Данный результат очевиден, так как если увеличивается объем, то возрастает количество мест, которое смогут занять частицы при неизменяющемся их количестве. Следовательно, растет число разных возможностей расположения на этих местах (увеличивается количество пространственных микросостояний). Увеличение числа микросостояний означает увеличение энтропии (см формулу (2)).
Изменение энтропии в изохорическом процессе
Рассмотрим изохорный процесс в идеальном газе ($V=const$ или $dV=0$). Из формулы (7) для изохорного процесса следует:
$dS=dleft( C_{V}lnT+RlnV right)=d(C_{V}lnT)left( 9 right)$.
После интегрирования (9), получим:
$S_{2}-S_{1}=C_{V}ln left( frac{T_{2}}{T_{1}} right)left( 10 right)$.
Формула (10) показывает, что в изохорическом процессе при увеличении температуры происходит рост энтропии. Данный результат можно пояснить так:
- при увеличении температуры растет средняя энергия частиц газа;
- увеличивается количество возможных энергетических состояний.
Изменение энтропии в адиабатном процессе
Адиабатный процесс характеризуется тем, что он происходит без теплообмена (δQ=0). Исследуя адиабатный процесс в идеальном газе, за основу для вычисления энтропии примем выражение (8). Найдем интеграл правой и левой частей этого выражения, получим:
$S_{2}-S_{1}=C_{V}ln left( frac{T_{2}}{T_{1}} right)+Rln {left( frac{V_{2}}{V_{1}} right)left( 11 right).}$
Запишем уравнение адиабатного процесса в параметрах $T,V$:
$T_{1}V_{1}^{gamma -1}=T_{2}V_{2}^{gamma -1}left( 12 right)$.,
где $gamma =frac{C_{p}}{C_{V}}-$ показатель адиабаты. Возьмем натуральные логарифмы от обеих частей выражения (12), имеем:
$ln left( frac{T_{2}}{T_{1}} right)=left( gamma -1 right)ln left( frac{V_{1}}{V_{2}} right)=-left( gamma -1 right)ln left( frac{V_{2}}{V_{1}} right)left( 13 right)$.
Преобразуем выражение (11), приняв во внимание формулу (13):
$S_{2}-S_{1}=C_{V}left[ -left( gamma -1 right)ln left(frac{V_{2}}{V_{1}} right) right]+Rln left( frac{V_{2}}{V_{1}} right)=-C_{V}left( frac{C_{p}}{C_{V}}-1 right)ln left( frac{V_{2}}{V_{1}}right)$
$+Rln {left( frac{V_{2}}{V_{1}} right)=left[ -C_{V}left(frac{C_{p}}{C_{V}}-1 right)+R right]ln {left( frac{V_{2}}{V_{1}}right)=left[ C_{V}-C_{p}+R right]ln left( frac{V_{2}}{V_{1}} right)}}left( 14 right)$.
Вспомним соотношение Майера:
$R=C_{p}-C_{V}left( 15 right)$
и сделаем вывод о том, что изменение энтропии в адиабатном процессе нет:
$S_{2}-S_{1}=0.$
Адиабатный процесс является изоэнтропийным ($S=const$).
При адиабатном расширении газа увеличение энтропии может идти только за счет увеличения объема, но при этом происходит уменьшение температуры, и энтропия уменьшается из-за уменьшения температуры. Данные тенденции взаимно компенсируют друг друга.
Рассмотрим пример. Два сосуда разного объема содержат по $nu$ молей одинакового идеального газа. Температуры газов равны $ T_1$ и $ T_2$. Сосуды соединяют, газы перемешиваются. Система приходит в состояние равновесия. Определим изменение энтропии в этом процессе.

Рисунок 1. Изменение энтропии в адиабатном процессе. Автор24 — интернет-биржа студенческих работ
Будем считать, что изобарное расширение каждого из газов до конечного объема является обратимым. В этом процессе температура каждой из газовых компонент изменяется до конечной температуры $frac{T_{1}+T_{2}}{2}$. Найдем изменение энтропии каждого из газов по отдельности:
$Delta S_{1}=intlimits_a^b frac{dT}{T} =nu c_{p}ln left(frac{T_{1}+T_{2}}{2T_{1}} right)left( 16 right)$,
где $a=T_{1};b=frac{T_{1}+T_{2}}{2}$ $c_p$ – молярная теплоемкость газа при постоянном давлении.
Изменение энтропии второго газа запишем аналогично:
$Delta S_{2}=nu c_{p}ln left( frac{T_{1}+T_{2}}{2T_{2}} right)left( 17right)$.
Поскольку энтропия является аддитивной величиной, то полное изменение энтропии найдем как сумму:
$Delta S_{2}=Delta S_{1}+Delta S_{2}=nu c_{p}ln left( frac{T_{1}+T_{2}}{2T_{1}} right)+c_{p}ln left(frac{T_{1}+T_{2}}{2T_{2}} right)=nu c_{p}ln frac{left( T_{1}+T_{2} right)^{2}}{4T_{1}T_{2}}=2nu$ $c_{p}lnleft( frac{T_{1}+T_{2}}{2sqrt {T_{1}T_{2}} } right)$.
Находи статьи и создавай свой список литературы по ГОСТу
Поиск по теме
| Энтропия | |
|---|---|
 |
|
| Размерность |
 |
| Единицы измерения | |
| СИ | Дж/К |
| СГС | эрг/К |
| Термодинамические величины |
|---|
 |
| Статья является частью серии «Термодинамика» |
См. также: Термодинамические потенциалы. |
| Разделы термодинамики |
|
| См. также «Физический портал» |
Термодинамическая энтропия 
Утверждение о существовании энтропии и перечисление её свойств составляют содержание второго и третьего начал термодинамики. Значимость данной величины для физики обусловлена тем, что наряду с температурой её используют для описания термических явлений и термических свойств макроскопических объектов. Качественные представления о термическом состоянии системы связаны с тепловыми ощущениями, выражаемыми понятиями «теплее», «холоднее», «нагрев», «охлаждение», «степень нагретости». К термическим относят свойства, характеризующие поведение вещества при его нагреве или охлаждении: термические коэффициенты, теплоёмкость и другие калорические коэффициенты, постоянную Кюри, показатели термостойкости, пределы огнестойкости и т. д.; примерами термических явлений служат термическое расширение, пироэлектричество, электрокалорический эффект, теплопроводность, изменение агрегатного состояния — кристаллизация и замерзание, плавление и таяние, испарение, кипение, сублимация (возгонка), конденсация и другие процессы.
Историческая справка[править | править код]
Понятие энтропии, её обозначение и название были введены Р. Клаузиусом (1865). Абстрактность этого понятия — одного из краеугольных камней термодинамики — и разнообразие подходов к обоснованию существования энтропии как термодинамической величины привели к появлению аксиоматики термодинамики.
Термодинамическая дефиниция энтропии[править | править код]
В термодинамике энтропию вводят, обосновывая её существование, перечисляя её свойства и строя для неё шкалу измерения на основании первого, второго и третьего начал термодинамики.
В термодинамических формализмах Клаузиуса[1] и Каратеодори[2] энтропию вводят одновременно с абсолютной термодинамической температурой. Математический аппарат термодинамики Гиббса[3] основан на использовании энтропии в качестве независимой термодинамической переменной, тогда как температура — естественный кандидат на эту роль, вводится как функция внутренней энергии и энтропии. Наконец, в рациональной термодинамике энтропию выражают через внутреннюю энергию и температуру, которые рассматривают как основные неопределяемые переменные теории.
Энтропия простой системы[править | править код]
Первое начало (закон) термодинамики устанавливает связь между внутренней энергией, работой и теплотой: одна из этих физических величин задаётся с помощью двух других, которые, будучи исходными объектами теории, в рамках самой этой теории определены быть не могут просто потому, что не существует понятий более общих, под которые их можно было бы подвести[4]. Термодинамика заимствует понятия энергии и работы из других разделов физики[5][6], тогда как определение количеству теплоты, наоборот, даётся только и именно в термодинамике. Согласно Клаузиусу теплоту 



|
(Дефиниция теплоты по Клаузиусу) |
Первое начало в этой формулировке вводит теплоту как физическую характеристику процесса, поведение которой определяется законом сохранения энергии, но не определяет её как математический объект. Детализировать дефиницию теплоты проще всего для равновесного процесса, когда работу, а следовательно и теплоту, можно выразить через переменные состояния. Для бесконечно малого[11] равновесного процесса в простой системе[12] возможен единственный вид работы — работа расширения/сжатия 

|
(Работа расширения/сжатия для равновесного процесса в простой системе) |
где 



|
(Первое начало для равновесного процесса в простой системе) |
где 
Это выражение, определяющее элементарную теплоту как математический объект, есть линейная дифференциальная форма (форма Пфаффа) для двух независимых переменных. Для данной пфаффовой формы условие интегрируемости Эйлера не выполняется, то есть 


|
(Дефиниция энтропии простой равновесной системы) |
где

есть интегрирующий делитель для формы Пфаффа. Клаузиус назвал функцию состояния 
Интегрирование уравнения для энтропии приводит к появлению в выражении для функции
Энтропия закрытой системы в термодинамике Клаузиуса — Каратеодори[править | править код]
Традиционный подход к построению термодинамики (аксиоматика Клаузиуса — Каратеодори) основан на использовании представления о внутренней энергии как базовом понятии теории, заимствовании формул для вычисления термодинамической работы из механики и электродинамики сплошных сред, и первом начале термодинамики в формулировке Клаузиуса.
Помимо работы расширения/сжатия система может одновременно выполнять другие виды работ, например работу по изменению площади поверхности раздела фаз, работу перемещения в поле тяготения, работу поляризации диэлектрика в электрическом поле и т. д. Объединяет все эти виды работ формальная структурная идентичность расчётных формул друг с другом и с выражением для работы расширения/сжатия[23][24][25]:

|
где 



Если однородная система одновременно совершает несколько различных видов работ, то они суммируются и полная работа системы 

|
(Суммарная работа для равновесного процесса в однородной системе) |
а для первого начала термодинамики получаем соотношение[30][31]:

|
(Первое начало для равновесного процесса в однородной системе) |
которое как и в случае простой системы представляет собой форму Пфаффа. Следствием второго начала термодинамики в любой его формулировке является вывод о том, что пфаффова форма

|
(Энтропия однородной закрытой системы) |
и абсолютная термодинамическая температура

Выражение 

Энтропия открытой системы[править | править код]
Принимая, что теплота и работа являются двумя единственно возможными формами передачи энергии[32][33][34], а изменение энергии, связанное с переносом вещества в открытой системе, есть составная часть общей работы, называемая химической работой (работой перераспределения масс веществ[35]), в случае однородной открытой системы дифференциальную форму 

|
(Химическая работа для равновесного процесса в открытой однородной системе) |
где 


Пфаффова форма

|
(Первое начало для равновесного процесса в открытой однородной системе) |
Дальнейшие рассуждения о существовании энтропии
и абсолютной термодинамической температуры

для открытой системы ничем не отличаются от соображений, высказанных при рассмотрении закрытой системы, поэтому ниже перечислены причины, по которым открытые системы потребовали отдельного рассмотрения.
Первая из этих причин состоит в том, что использование в понятийном аппарате термодинамики химической работы как части общей работы делает неэквивалентными представления об адиабатной изоляции как накладывающей запрет на обмен веществом (то есть любая адиабатно изолированная система есть система закрытая или, говоря иначе, масса есть адиабатически заторможенная величина)[42][43][44][45], и адиабатной изоляции как допускающей обмен энергией только в форме работы[46][47]. Восстановить эквивалентность приведённых выше формулировок об адиабатной изоляции удаётся, если модифицировать дефиницию теплоты по Клаузиусу, добавив к теплоте и работе третью форму передачи энергии — энергию переноса массы 

|
(Модифицированная дефиниция теплоты для открытой системы) |
где
Вторая причина отдельного рассмотрения вопроса об энтропии открытых систем заключается в следующем. За исключением химического потенциала все входящие в пфаффову форму

|
(Энтропия открытой однородной системы по Гиббсу; фундаментальное уравнение Гиббса в энтропийном выражении) |
как функции состояния, при неизменности масс компонентов совпадающей с энтропией однородной закрытой системы. Из фундаментального уравнения Гиббса в дифференциальной форме[55]

|
(Дифференциальная форма фундаментального уравнения Гиббса для энтропии) |
находим значения частных производных энтропии:



Химический потенциал 

|
(Дефиниция химического потенциала компонента) |
Энтропия в термодинамике Гиббса[править | править код]
Построение теории на основе постулирования существования энтропии как функции состояния, в состав независимых переменных которой входят массы компонентов, составляет главное содержание термодинамики Гиббса[57], а способ, каким выполнено распространение термодинамики Клаузиуса на открытые системы, позволяет говорить об аксиоматике Гиббса[52][45]. В термодинамике Гиббса вводят понятия компонента системы, фазы и многофазной гетерогенной системы, постулируют существование внутренней энергии 
Обратите внимание, что содержательную дефиницию температуры по Гиббсу[59][60][61]

|
(Термодинамическая температура по Гиббсу) |
можно, с другой стороны, рассматривать и как описательную дефиницию энтропии. А именно, энтропия в термодинамике Гиббса есть такая экстенсивная переменная состояния, что производная внутренней энергии по энтропии представляет собой интенсивную переменную состояния, обладающую всеми положенными термодинамической температуре свойствами.
Энтропия в рациональной термодинамике[править | править код]
Рациональная термодинамика не подразделяет термодинамику на равновесную и неравновесную; обе эти дисциплины рассматриваются как единая часть физики сплошных сред[62][63][64][65]. Равновесная рациональная термодинамика есть результат применения общей теории к системам в состоянии равновесия[66]. Исходные неопределяемые понятия теории — энергия 


при механическом равновесии имеющем одинаковое значение во всех частях системы, вводят химический потенциал

как интенсивную величину, имеющую при химическом равновесии одно и то же значение во всех частях системы[67].
Абсолютную термодинамическую температуру вводят посредством следующей аксиомы: существует интенсивная термодинамическая величина, температура

которая характеризует степень нагретости тел и обладает следующими свойствами[68]:
- в выбранном за начало отсчёта состоянии температура равна нулю
;
- температура монотонно растёт с увеличением энергии системы
;
- при термодинамическом равновесии имеет одно и то же значение во всех частях системы.
Энтропию в рациональной термодинамике задают как аддитивную величину, равную[69]

Свойства энтропии, вытекающие из этого определения[69]:
;
;
;
где 
Свойства энтропии[править | править код]
Перечисление свойств энтропии дано применительно к термодинамике Гиббса; примеры, приводимые для иллюстрации перечисляемых свойств энтропии, относятся, как правило, к открытым однородным термодеформационным системам, для которых справедливо фундаментальное уравнение Гиббса в энтропийном выражении[70][71]:

|
(Фундаментальное уравнение Гиббса в энтропийном выражении для открытой термодеформационной системы) |
- Энтропия в фундаментальном уравнении в энтропийном выражении есть однозначная дифференцируемая функция аддитивных независимых переменных[72][44][73] с непрерывными первыми и кусочно-непрерывными вторыми производными.
- Энтропия есть величина аддитивная[74][75][76][72][77][78][79][80], то есть энтропия термодинамической системы равна сумме энтропий всех её частей. Аддитивность энтропии позволяет распространить это понятие на термодинамические системы любой сложности.
- Как следствие аддитивности получаем, что энтропия в фундаментальном уравнении в энтропийном выражении есть однородная функция первого порядка всех независимых переменных[81][73], то есть для


|
- и для неё справедливо тождество (теорема) Эйлера[82]:

|
- Для однородной системы частная производная энтропии по внутренней энергии есть величина, обратная абсолютной термодинамической температуре (термодинамическая дефиниция температуры как следствие второго начала термодинамики)[83][60][61][84]:
![Tequiv left[left({frac {partial S}{partial U}}right)_{{V,{x_{i}}}}right]^{{-1}}.](https://wikimedia.org/api/rest_v1/media/math/render/svg/0befaea6b14739d02ea258e6fd840d6e6790518b)
|
(Термодинамическая дефиниция температуры) |
- В соответствии с теоремой об обратных величинах[85] это определение совпадает с дефиницией равновесной температурой по Гиббсу[59][60][61]:

|
(Термодинамическая температура по Гиббсу) |
- Энтропия есть монотонная функция внутренней энергии
- Энтропия есть величина полуограниченная. Традиционно принимают, что энтропия ограничена снизу, то есть для каждой термодинамической системы существует состояние с наименьшей энтропией[88].
- Температура есть величина полуограниченная. Абсолютная температурная шкала Кельвина построена так, что

- Нуль и бесконечность допустимы в качестве пределов[88].
- Для любой термодинамической системы состояния с наименьшей энтропией и наименьшей температурой совпадают (постулат Планка)[88][89]. С приближением температуры к абсолютному нулю энтропия перестаёт зависеть от температуры и приближается к определённому постоянному значению, которое можно положить равным нулю[90] и принять за начало отсчёта энтропии, устранив тем самым упомянутый в разделе Энтропия простой системы произвол в выборе постоянной интегрирования для энтропии:

|
(Третье начало термодинамики; тепловая теорема Нернста) |
- Согласно постулату Тиссы внутренняя энергия ограничена и эта граница соответствует абсолютному нулю температуры[88][91]. Таким образом, состояние системы при абсолютном нуле температуры, когда все термодинамические величины, характеризующие равновесное состояние, перестают зависеть от температуры[92], наилучшим образом подходит в качестве стандартного состояния начала отсчёта основных термодинамических величин.
- Энтропия изолированной системы в состоянии термодинамического равновесия имеет максимальное значение (постулат Гиббса)[93][92], то есть для равновесия изолированной системы необходимо и достаточно, чтобы при всех возможных (не нарушающих постоянства внутренней энергии, обобщённых координат и масс компонентов) изменениях её состояния вариация энтропии
системы не была положительной[94]:

|
(Условие равновесия изолированной системы) |
- Поскольку речь идёт об изолированной системе, внешнее воздействие на которую запрещено, понятие вариации в данном случае означает виртуальное изменение энтропии[95]. Знак равенства в этом выражении относится к безразличному равновесию.
- Условие равновесия Гиббса вытекает из входящего в состав второго начала термодинамики постулата Клаузиуса о неубывании энтропии адиабатно изолированной системы[94]
- Энтропия в фундаментальном уравнении в энтропийном выражении есть характеристическая функция, то есть фундаментальное уравнение содержит все термодинамические сведения о данной системе[73].
Энтропия как характеристическая функция[править | править код]
Энтропия и теплота квазистатического (равновесного) процесса[править | править код]
Из выражения для первого начала в открытой однородной системе и дифференциальной формы фундаментального уравнения Гиббса для энтропии получаем выражение для элементарной теплоты равновесного (квазистатического) процесса[96][97]:

|
(Теплота элементарного равновесного процесса) |
(для простой равновесной системы это выражение непосредственно вытекает из дефиниции энтропии).
Данное соотношение, связывающее термодинамику Клаузиуса с термодинамикой Гиббса, представляет интерес для пользователей, которым требуется изложить материал из старой учебной и научной литературы с применением терминологии, либо вовсе не использующей понятие «теплота», либо использующей его как определяемое через энтропию и абсолютную температуру вторичное понятие.
Энтропия как характеристика изотермического процесса[править | править код]

Зависимость удельной энтропии воды от температуры
Для равновесного изотермического процесса с нулевой работой интегрирование выражения для теплоты равновесного процесса даёт следующее выражение для изменения внутренней энергии:

то есть в любом равновесном изотермическом процессе с нулевой работой энергия расходуется на увеличение энтропии системы и выделяется при уменьшении энтропии. Преобразуем это уравнение к виду

и назовём отношение 
Рассмотрим в качестве примера фазовые переходы в воде при атмосферном давлении (см. рисунок). При таянии льда подводимая к системе энергия расходуется на увеличение энтропии системы вследствие изменения структуры H2O, тогда как температура системы лёд + вода остаётся близкой к 0 °C (273 К) до полного исчезновения льда. При замерзании воды имеет место обратная ситуация: энергия выделяется в окружающую среду при 0 °C. Нагрев воды, образовавшейся при таянии льда, ведёт к повышению температуры воды вплоть до её закипания при 100 °C (373 К). Кипение воды при постоянном давлении есть процесс изотермический: подводимая энергия расходуется на испарение воды и увеличение энтропии системы вода + водяной пар, тогда как температура остаётся близкой к 100 °C до полного исчезновения жидкой воды.

Статистическое определение энтропии: принцип Больцмана[править | править код]
В 1877 году Людвиг Больцман установил связь энтропии с вероятностью данного состояния. Позднее эту связь представил в виде формулы Макс Планк:

где константа 

Рассмотрим, например, идеальный газ в сосуде. Микросостояние определено как позиции и импульсы (моменты движения) каждого составляющего систему атома. Связность предъявляет к нам требования рассматривать только те микросостояния, для которых:
(I) месторасположения всех частей расположены в рамках сосуда,
(II) для получения общей энергии газа кинетические энергии атомов суммируются.
Согласно определению, энтропия является функцией состояния, то есть не зависит от способа достижения
этого состояния, а определяется параметрами этого состояния. Так как
Понимание энтропии как меры беспорядка[править | править код]
Существует мнение, что мы можем смотреть на энтропию и как на меру беспорядка в системе. В определённом смысле это может быть оправдано, потому что мы думаем об «упорядоченных» системах как о системах, имеющих очень малую возможность конфигурирования, а о «беспорядочных» системах как об имеющих очень много возможных состояний. Собственно, это просто переформулированное определение энтропии как числа микросостояний на данное макросостояние.
Рассмотрим, например, распределение молекул идеального газа. В случае идеального газа наиболее вероятным состоянием, соответствующим максимуму энтропии, будет равномерное распределение молекул. При этом реализуется и максимальный «беспорядок», так как при этом будут максимальные возможности конфигурирования.
Получившее повсеместное распространение понимание энтропии как меры беспорядка в термодинамической системе не является, тем не менее, общепринятым[98]: «Тождественность энтропии с беспорядком не только никем никогда не была доказана и не только не может быть доказана в принципе, но и прямо противоречит реально наблюдаемым фактам…»[98]; «…применительно к реальным системам энтропия не является мерой беспорядка»[99]; «…в ходе роста энтропии Вселенной общая её (Вселенной) сложность растёт, однако для составляющих Вселенную реальных (под)систем энтропия мерой беспорядка/сложности не является»[100].
Границы применимости понимания энтропии как меры беспорядка[править | править код]
Подобное определение беспорядка термодинамической системы как количества возможностей конфигурирования системы фактически дословно соответствует определению энтропии как числа микросостояний на данное макросостояние. Проблемы начинаются в двух случаях:
- когда начинают смешивать различные понимания беспорядка, и энтропия становится мерой беспорядка вообще;
- когда понятие энтропии применяется для систем, не являющихся термодинамическими.
В обоих этих случаях применение понятия термодинамической энтропии совершенно неправомерно[101].
Рассмотрим оба пункта подробнее.
Рассмотрим пример термодинамической системы — распределение молекул в поле тяготения. В этом случае наиболее вероятным распределением молекул будет распределение согласно барометрической формуле Больцмана. Другой пример — учёт электромагнитных сил взаимодействия между ионами. В этом случае наиболее вероятным состоянием, соответствующим минимуму свободной энергии, будет упорядоченное кристаллическое состояние, а совсем не «хаос», хотя в состоянии «хаоса» значение конфигурационной энтропии системы и ниже. (Термин «хаос» здесь понимается в смысле беспорядка — в наивном смысле. К хаосу в математическом смысле как сильно неустойчивой нелинейной системе это не имеет отношения, конечно.)
Рассмотрим случай с кристаллической решёткой более подробно. Кристаллическая решётка может быть и в равновесном, и в неравновесном состоянии, как и любая термодинамическая система. Скажем, возьмём следующую модель — совокупность взаимодействующих осцилляторов. Рассмотрим некоторое неравновесное состояние: все осцилляторы имеют одинаковое отклонение от положения равновесия. С течением времени эта система перейдёт в состояние ТД равновесия, в котором отклонения (в каждый момент времени) будут подчинены некоторому распределению типа Максвелла (только это распределение будет для отклонений, и оно будет зависеть от типа взаимодействия осцилляторов). В таком случае максимум энтропии будет действительно реализовывать максимум возможностей конфигурирования, то есть — беспорядок согласно вышеуказанному определению. Но данный «беспорядок» вовсе не соответствует «беспорядку» в каком-либо другом понимании, например, информационному. Такая же ситуация возникает и в примере с кристаллизацией переохлаждённой жидкости, в которой образование структур из «хаотичной» жидкости идёт параллельно с увеличением энтропии.
То есть при образовании кристалла из переохлаждённой жидкости энтропия увеличивается с одновременным ростом температуры. Если кристаллизация сопровождается отводом тепла из системы, то энтропия при этом уменьшится.
Это неверное понимание энтропии появилось во время развития теории информации, в связи с парадоксом термодинамики, связанным с мысленным экспериментом так называемый «демона Максвелла». Суть парадокса заключалась в том, что рассматривалось два сосуда с разными температурами, соединённых узкой трубкой с затворками, которыми управлял так называемый «демон». «Демон» мог измерять скорость отдельных летящих молекул, и таким образом избирательно пропускать более быстрые в сосуд с высокой температурой, а более медленные — в сосуд с низкой. Из этого мысленного эксперимента вытекало кажущееся противоречие со вторым началом термодинамики.
Парадокс может быть разрешён при помощи теории информации. Для измерения скорости молекулы «демон» должен был бы получить информацию о её скорости. Но всякое получение информации — материальный процесс, сопровождающийся возрастанием энтропии. Количественный анализ[102] показал, что приращение энтропии при измерении превосходит по абсолютной величине уменьшение энтропии, вызванное перераспределением молекул «демоном».
Измерение энтропии[править | править код]
В реальных экспериментах очень трудно измерить энтропию системы. Техники измерения базируются на термодинамическом определении энтропии и требуют экстремально аккуратной калориметрии.
Для упрощения мы будем исследовать механическую систему, термодинамические состояния которой будут определены через её объём 

где нижний индекс 

Таким образом, мы можем получить значение энтропии любого состояния (


В добавление, если путь между первым и последним состояниями лежит сквозь любой фазовый переход первого рода, скрытая теплота, ассоциированная с переходом, должна также учитываться.
Энтропия первоначального состояния должна быть определена независимо. В идеальном варианте выбирается первоначальное состояние как состояние при экстремально высокой температуре, при которой система существует в виде газа. Энтропия в этом состоянии подобна энтропии классического идеального газа плюс взнос от молекулярных вращений и колебаний, которые могут быть определены спектроскопически.
Построение графика изменения энтропии[править | править код]
Следующее уравнение может быть использовано для построения графика изменения энтропии на диаграмме 

Здесь два замечания:
«Что такое энтропия?»[править | править код]

Что сложнее: паровоз или лошадь? Когда появились первые паровозы, путеец рассказал обратившимся к нему ошеломлённым крестьянам об устройстве и действии паровой машины и закончил объяснение вопросом: «Всё понятно?» — «Всё! Кроме того, где же внутри находится лошадь?» Крестьяне с детства знают лошадь, она для них в объяснении не нуждается. С научной точки зрения лошадь несравненно более сложна, чем тепловая машина, так что наука пыталась представить лошадь как тепловую машину, а не наоборот
Однозначного ответа на этот вопрос не существует по той простой причине, что разных энтропий много — представление об энтропии используется в различных научных дисциплинах: термодинамике, статистической физике, теории информации и др. Но и внутри каждой из перечисленных дисциплин единообразия также нет и в помине: в теории информации рассматривают энтропию Шеннона, энтропию Реньи, энтропию Чисара, энтропию Хаврда — Чарват — Дароши[103]; статистическая физика оперирует энтропиями Больцмана, Гиббса, Цаллиса; существуют различные дефиниции термодинамической энтропии. Содержательная дефиниция той или иной конкретной энтропии зависит от аксиоматики системы построения/изложения, использующей эту энтропию. По указанной причине не существует универсальной дефиниции термодинамической энтропии, ибо для различных аксиоматических систем термодинамики ответ на вынесенный в заголовок вопрос будет различен.
Студенты, приступающие к изучению термодинамики, часто жалуются на непонятность энтропии, связанную с отсутствием наглядности (рисунок иллюстрирует относительность представлений людей о наглядности, понятности и простоте термодинамических систем[K 1]).
Для описания термических явлений в физике вводят новые комплементарные (взаимодополняющие) физические величины — температуру и энтропию, — содержательные дефиниции которым не дают ни в механике, ни в электродинамике. В термодинамике Клаузиуса — Каратеодори энтропию вводят как приведённую внутреннюю энергию изотермической системы, то есть разность энтропий равна приведённой теплоте изотермического процесса.
В термодинамике Гиббса и в системе А. А. Гухмана энтропия представляет собой неопределяемое базовое понятие — таков в этих системах содержательный ответ на рассматриваемый вопрос[K 2]. В термодинамической аксиоматике А. А. Гухмана [105][106][107][108][109][110] и рациональной термодинамике в трактовке П. А. Жилина[65][K 3] и температуру, и энтропию вводят как неопределяемые базовые переменные, несводимые к более простым[K 4]. Пояснительное дополнение к дефиниции энтропии по Гухману разъясняет, что энтропия есть координата состояния при термическом взаимодействии (теплообмене), которое выражается в переносе энтропии от одного объекта к другому[112].
В рациональной термодинамике в трактовке школы Трусделла в качестве базовой термической переменной используют температуру

но, в отличие от другой функции энергии и температуры, — теплоёмкости 

в формулу, служащую дефиницией энтропии, входит не производная, а интеграл. Пояснительное дополнение к ответу, пригодное почти[K 5] для любого способа изложения термодинамики, сообщает, что энтропия необходима для построения математического аппарата термодинамики и, следовательно, привлечение термодинамики к решению любой научной или практической задачи явно или неявно подразумевает использование энтропии. Польза, приносимая людям от обращения к термодинамике, есть польза от ввода энтропии в понятийный аппарат науки. Дополнительно к сказанному можно провести аналогию с теплоёмкостью: если для неизотермических процессов нагрева льда, жидкой воды и водяного пара затраты энергии равны произведению теплоёмкости на разность температур, то для изотермических процессов таяния и кипения затраты энергии равны произведению температуры на разность энтропий.
Понять энтропию и её значение для термодинамики означает, что необходимо знать происхождение этой величины, понимать её связи с другими термодинамическими переменными и уметь применять энтропию на практике[K 6][104].
См. также[править | править код]
- Аксиоматика термодинамики
- Внутренняя энергия
- Демон Максвелла
- Негэнтропия
- Стрела времени
- Тепловая смерть
- Температура
- Термодинамика чёрных дыр
- Характеристическая функция
- Энтропия
- Энтропия Гиббса
- Энтропия Реньи
- Энтропия Цаллиса
- Энтропия Чисара
- Энтропия Шеннона
Комментарии[править | править код]
- ↑ Наглядность, понятность, очевидность и простота есть суждения относительные, зависящее как от обыденности понятия, так и от уровня знаний человека. Крестьяне с детства знали лошадь, и она для них наглядна и понятна. Для теплотехников наглядна и понятна тепловая машина, а не лошадь. В. Томсон как-то на лекции спросил студентов: «Знаете ли вы, кто такой математик?» Написав на аудиторной доске:
, Томсон повернулся к студентам и, указывая на эту формулу, сказал: «Математик — тот, для кого это так же очевидно, как для вас то, что дважды два — четыре»[104].
- ↑ Описательная характеристика энтропии как термической (тепловой) координаты состояния не отменяет того факта, что в системе Гухмана энтропия входит в число основных неопределяемых понятий теории.
- ↑ Вот цитата из статьи К. Трусделла, демонстрирующая совпадение его взглядов с подходом П. А. Жилина: «Я повторяю в течение уже многих лет, пренебрегая насмешками людей, наделённых физической интуицией, что температура и энтропия являются наряду с массой, положением и временем первоначальными неопределяемыми переменными. Они описываются только такими свойствами, которые можно выразить языком математики»[111]. Эти воззрения на температуру и энтропию отличаются от тех, которые сейчас принято рассматривать как отличительную особенность «рациональной термодинамике в трактовке школы Трусделла».
- ↑ Чтобы дать содержательную дефиницию какому-либо понятию, нужно указать, частным случаем какого более общего понятия оно является. Если более фундаментального понятия не существует, то понятие в конце цепочки дефиниций является неопределяемым — базовым (первичным, исходным, начальным) понятием аксиоматической системы, несводимым к более простым. В любой науке имеются такие первичные понятия, те элементарные кирпичики, из которых строятся все остальные, производные понятия, и которым не даются содержательные дефиниции в самой научной дисциплине. Примерами неопределяемых базовых понятий служат: в математике — множество, в физике — пространство, время, масса, энергия и др. Невозможность дать понятию или переменной содержательной дефиниции без выхода за границы изучаемой дисциплины, во-первых, не означает запрета на использование для базового понятия/переменной описательных дефиниций, и, во-вторых, свойства базовых понятий/переменных описываются аксиомами рассматриваемой теории. Иными словами, набор базовых понятий/переменных научной дисциплины зависит от выбора системы изложения/построения этой дисциплины, а полный набор её аксиом образует систему содержательных дефиниций базовых понятий/переменных теории.
- ↑ Слово «почти» служит напоминанием о том, что любую систему построения/изложения термодинамики, в которой энтропия есть понятие вторичное (выводимое из понятий более общих), можно в принципе преобразовать в другую систему — «безэнтропийную термодинамику», — в которой энтропию как понятие необязательное уже не используют[115]. В связи со сказанным упомянем, что автор третьего закона термодинамики Вальтер Нернст в своих работах не «пользовался понятием энтропии, которое он считал неясным и потому попросту не любил его»[116] (Нернст использовал свободную энергию Гельмгольца, связанную с максимальной работой, то есть энтропию он заменял взятой со знаком «минус» производной от энергии Гельмгольца по температуре[117]).
- ↑ В связи со сказанным представляют интерес воспоминания И. К. Кикоина, посещавшего в студенческие годы семинар В. А. Фока и рассказавшего историю про поиск решения сложной задачи по электростатике: «…в конце концов, получили длиннющее дифференциальное уравнение. Оно занимало всю доску. За математическими выкладками мы следили очень внимательно, так что с математикой всё было в порядке, а вот усмотреть физический смысл, скрытый за этой длинной формулой, мы не могли. Кто-то из студентов спросил Владимира Александровича: “А какой физический смысл имеет это уравнение?”. — Он на нас посмотрел с укором и сказал: “А физический смысл этого уравнения заключается в том, что оно имеет решение”»[118].
Примечания[править | править код]
- ↑ Клаузиус, 1934.
- ↑ Каратеодори.
- ↑ Гиббс Дж. В., Термодинамика. Статистическая механика, 1982.
- ↑ Хазен, 2000, глава VI, раздел 4. Полная и замкнутая формулировка аксиоматического определения энтропии и начал термодинамики..
- ↑ Петров Н., Бранков Й., Современные проблемы термодинамики, 1986, с. 35.
- ↑ Семенченко, 1966, с. 54.
- ↑ Clausius, 1887, S. 33.
- ↑ Борн, 1964, с. 230–231.
- ↑ Знак
или
перед
- ↑ Борн, 1964, с. 231.
- ↑ Элементарным (инфинитезимальным) называют процесс, для которого разница между начальным и конечным состояниями системы бесконечно мала.
- ↑ Простой называют закрытую термодеформационную систему, представляющую собой однородную изотропную среду (фазу) неизменного химического состава и массы, описываемую посредством переменных
- ↑ Сивухин Д. В., Термодинамика и молекулярная физика, 2005, с. 59.
- ↑ 1 2 Радушкевич Л. В., Курс термодинамики, 1971, с. 36.
- ↑ Базаров И. П., Термодинамика, 2010, с. 37.
- ↑ Условия интегрируемости дифференциальных полиномов подробно рассмотрены в книге Белоконь Н. И., Термодинамика, 1954, с. 137—138.
- ↑ Кричевский И. Р., Понятия и основы термодинамики, 1970, с. 270.
- ↑ Сычёв, 1991, с. 22.
- ↑ 1 2 Путилов К. А., Термодинамика, 1971, с. 13.
- ↑ Квасников, 2002, с. 43.
- ↑ 1 2 Сычёв, 1991, с. 24.
- ↑ Радушкевич Л. В., Курс термодинамики, 1971, с. 111.
- ↑ 1 2 Базаров И. П., Термодинамика, 2010, с. 27.
- ↑ Сычёв, 2009, с. 13.
- ↑ Кубо Р., Термодинамика, 1970, с. 21.
- ↑ Базаров И. П., Термодинамика, 2010, с. 27–29.
- ↑ Семенченко, 1966, с. 55.
- ↑ Сычёв, 2009, с. 14.
- ↑ 1 2 Кубо Р., Термодинамика, 1970, с. 20.
- ↑ Базаров И. П., Термодинамика, 2010, с. 38.
- ↑ Радушкевич Л. В., Курс термодинамики, 1971, с. 38.
- ↑ Глазов В. М., Основы физической химии, 1981, с. 29.
- ↑ Путилов К. А., Термодинамика, 1971, с. 40.
- ↑ Сейдж Б. Х., Термодинамика многокомпонентных систем, 1969, с. 54.
- ↑ Тамм М. Е., Третьяков Ю. Д., Физико-химические основы неорганической химии, 2004, с. 11.
- ↑ Lebon, 2008, p. 14.
- ↑ Жариков, 2005, уравнение (2.4)..
- ↑ Callen, 1985, p. 36.
- ↑ Сычёв, 2009, с. 257.
- ↑ Путилов К. А., Термодинамика, 1971, с. 125.
- ↑ Использование масс компонентов, а не масс составляющих систему веществ в качестве обобщённых координат в выражении для химической работы означает отказ от прямого рассмотрения влияния химических реакций на массы веществ, ибо химические превращения в системе уже учтены при подсчёте числа компонентов.
- ↑ Квасников И. А., Молекулярная физика, 2009, с. 31.
- ↑ Квасников, 2002, с. 22.
- ↑ 1 2 Петров Н., Бранков Й., Современные проблемы термодинамики, 1986, с. 66.
- ↑ 1 2 3 Tisza, 1966.
- ↑ Бэр Г. Д., Техническая термодинамика, 1977, с. 73.
- ↑ Залевски К., Феноменологическая и статистическая термодинамика, 1973, с. 10.
- ↑ Пригожин И., Кондепуди Д., Современная термодинамика, 2002, с. 52.
- ↑ Кубо Р., Термодинамика, 1970, с. 16.
- ↑ Жилин П. А., Рациональная механика сплошных сред, 2012.
- ↑ «…Понятия энергии, температуры, энтропии и химического потенциала вводятся одновременно и по отдельности определить их принципиально нельзя» (с. 48), «…Нельзя сначала определить внутреннюю энергию, а затем химический потенциал и энтропию. Все эти понятия могут быть введены только одновременно» (с. 140).
- ↑ 1 2 Петров Н., Бранков Й., Современные проблемы термодинамики, 1986, с. 43.
- ↑ Воронин Г. Ф., Основы термодинамики, 1987, с. 50.
- ↑ Callen, 1985, p. 28—29.
- ↑ 1 2 Мюнстер А., Химическая термодинамика, 1971, с. 69.
- ↑ Мюнстер А., Химическая термодинамика, 1971, с. 70.
- ↑ Мюнстер А., Химическая термодинамика, 1971, с. 67.
- ↑ Сам Гиббс эти постулаты в своей основной термодинамической работе «О равновесии гетерогенных веществ» формулировал по мере необходимости, как бы мимоходом, и не называл принимаемые им без доказательства утверждения ни аксиомами, ни постулатами.
- ↑ 1 2 Гиббс Дж. В., Термодинамика. Статистическая механика, 1982, с. 93.
- ↑ 1 2 3 Guggenheim, 1985, p. 15.
- ↑ 1 2 3 Callen, 1985, p. 35.
- ↑ Трусделл К., Термодинамика для начинающих, 1970.
- ↑ Трусделл К., Первоначальный курс рациональной механики сплошных сред, 1975.
- ↑ Truesdell, 1984, 1984.
- ↑ 1 2 Жилин П. А., Рациональная механика сплошных сред, 2012.
- ↑ Максимов, 2009.
- ↑ Максимов, 2009, с. 5.
- ↑ Максимов, 2009, с. 5—6.
- ↑ 1 2 3 Максимов, 2009, с. 7.
- ↑ 1 2 Петров Н., Бранков Й., Современные проблемы термодинамики, 1986, с. 71–72.
- ↑ Мюнстер А., Химическая термодинамика, 1971, с. 90–91.
- ↑ 1 2 Callen, 1985, p. 28.
- ↑ 1 2 3 Мюнстер А., Химическая термодинамика, 1971, с. 92.
- ↑ Сорокин В. С., Макроскопическая необратимость и энтропия. Введение в термодинамику, 2004, с. 55.
- ↑ Воронин Г. Ф., Основы термодинамики, 1987, с. 51.
- ↑ Петров Н., Бранков Й., Современные проблемы термодинамики, 1986, с. 67.
- ↑ Кубо Р., Термодинамика, 1970, с. 85—86.
- ↑ Buchdahl H. A., The Concepts of Classical Thermodynamics, 1966, p. 74.
- ↑ Зоммерфельд А., Термодинамика и статистическая физика, 1955, с. 52.
- ↑ В термодинамике различают аддитивность по размерам системы (длине упругого стержня или пружины, площади поверхности раздела, объёму) и аддитивность по массе. Ясно, что последнее понятие не универсально, и даже аддитивность экстенсивных переменных по объёму не гарантирует, что к этим переменным применимо представление об аддитивности по массе. Например, оно непригодно для аддитивных по объёму переменных фотонного газа — системы с нулевой массой.
- ↑ Петров Н., Бранков Й., Современные проблемы термодинамики, 1986, с. 62.
- ↑ Тер Хаар Д., Вергеланд Г., Элементарная термодинамика, 1968, с. 117.
- ↑ Ландау Л. Д., Лифшиц Е. М., Статистическая физика. Часть 1, 2002, с. 51.
- ↑ Falk, Jung, 1959, S. 156.
- ↑ Сычёв, 1991, с. 15.
- ↑ Воронин Г. Ф., Основы термодинамики, 1987, с. 53.
- ↑ Отрицательная температура. БСЭ, 3-е изд., 1975, т. 19.
- ↑ 1 2 3 4 Петров Н., Бранков Й., Современные проблемы термодинамики, 1986, с. 68.
- ↑ Callen, 1985, p. 30.
- ↑ Базаров И. П., Термодинамика, 2010, с. 92.
- ↑ Tisza, 1966, p. 125.
- ↑ 1 2 Новиков И. И., Термодинамика, 1984, с. 106.
- ↑ Базаров И. П., Термодинамика, 2010, с. 121.
- ↑ 1 2 Воронин Г. Ф., Основы термодинамики, 1987, с. 102.
- ↑ Мысленное допустимое (не противоречащее условиям существования системы) изменение энтропии, не зависящее от времени. Встречающееся в литературе определение вариации как отклонения от равновесия, допускаемого наложенными на систему связями (условиями), означает то же самое.
- ↑ Базаров И. П. и др., Термодинамика и статистическая физика, 1986, с. 26.
- ↑ Базаров И. П., Термодинамика, 2010, с. 29.
- ↑ 1 2 Хайтун С. Д., Кризис теории познания, 2014, с. 98.
- ↑ Хайтун С. Д., Кризис теории познания, 2014, с. 100.
- ↑ Хайтун С. Д., Кризис теории познания, 2014, с. 102.
- ↑ Lambert Frank L. A Brief Introduction to the Second Law and to Entropy for Chemistry Students
- ↑ Бриллюэн Л. Наука и теория информации. — М., 1960.
- ↑ Зарипов Р. Г., Новые меры и методы в теории информации, 2005, с. 175.
- ↑ 1 2 Кричевский И. Р., Петрянов И. В. Термодинамика для многих, 1975, с. 146.
- ↑ Гухман, 2010, с. 11.
- ↑ Ляшков В. И., Теоретические основы теплотехники, 2015, с. 10.
- ↑ Цирлин А. М., Методы оптимизации в необратимой термодинамике и микроэкономике, 2003, с. 19.
- ↑ Исаев С. И., Курс химической термодинамики, 1986, с. 18.
- ↑ Жуковский В. С., Термодинамика, 1983, с. 11.
- ↑ Леонова В. Ф., Термодинамика, 1968, с. 19—20.
- ↑ Трусделл К., Термодинамика для начинающих, 1970, с. 117.
- ↑ Свиридов В. В., Свиридов А. В., Физическая химия, 2016, с. 113.
- ↑ Максимов, 2009, с. 7.
- ↑ Максимов, 2009, с. 6.
- ↑ Игнатович В. Н., Введение в диалектико-материалистическое естествознание, 2007, с. 411.
- ↑ Гельфер Я. М., История и методология термодинамики и статистической физики, 1981, с. 228.
- ↑ Nernst Walther, Theoretische Chemie, 1900, S. 29.
- ↑ Кикоин И. К. Рассказы о физике и физиках, 1986, с. 33.
Литература[править | править код]
- Buchdahl H. A. The Concepts of Classical Thermodynamics. — Cambridge: Cambridge University Press, 1966. — XI + 223 p.
- Callen H. B. Thermodynamics and an Introduction to Thermostatistics. — 2nd ed. — N. Y. e. a.: John Wiley, 1985. — xvi + 493 p. — ISBN 0471862568, 9780471862567.
- Clausius R. Die mechanische Wärmetheorie. Band 1. — 3 Auflage. — Braunschweig: Druck und Verlag von Friedrich Vieweg und Sohn, 1887. — XVI + 403 p.
- Ehrenfest-Afanassjewa T. Zur Axiomatisierung des zweiten Hauptsatzes der Thermodynamik (нем.) // Zeitschrift für Physik. — 1925. — Vol. 33, Nr. 1. — P. 933–945.
- Ehrenfest-Afanassjewa T. Berichtigung zu der Arbeit: Zur Axiomatisierung des zweiten Hauptsatzes der Thermodynamik (нем.) // Zeitschrift für Physik. — 1925. — Vol. 34, Nr. 1. — P. 638.
- Ehrenfest-Afanassjewa T. Die Grundlagen der Thermodynamik. — Leiden: E.J. Brill, 1956. — XII + 131 с.
- Falk G., Jung H. Axiomatik der Thermodynamik (нем.) // Flügge S. (ed.). Encyclopedia of Physics / Flügge S. (Hrsg.). Handbuch der Physik. — Springer-Verlag, 1959. — Vol. III/2. Principles of Thermodynamics and Statistics / Band III/2. Prinzipien der Thermodynamik und Statistik, S. 119–175.
- Guggenheim E. A. Thermodynamics: An Advanced Treatment for Chemists and Physicists. — 7th ed. — Amsterdam: North-Holland, 1985. — xxiv + 390 p. — ISBN 0 444 86951 4.
- Lebon G., Jou D., Casas-Vázquez J. Understanding Non-equilibrium Thermodynamics: Foundations, Applications, Frontiers. — Berlin — Heidelberg: Springer, 2008. — xiii + 325 p. — ISBN 978-3-540-74251-7, 978-3-540-74252-4. — doi:10.1007/978-3-540-74252-4.
- Nernst Walther. Theoretische Chemie vom Standpunkte der avogadro’schen Regel und der Thermodynamik. — Dritte Auflage. — Stuttgart: Verlag von Ferdinand Enke, 1900. — xiv + 710 p.
- Noll W. The Foundations of Mechanics and Thermodynamics: Selected Papers. — Berlin — Heidelberg — New York: Springer-Verlag, 1974. — X + 324 p. — ISBN 978-3-642-65819-8.
- Reif F. Fundamentals of statistical and thermal physics. — McGraw-Hill, 1965.
- Tisza Laszlo. Generalized Thermodynamics. — Cambridge (Massachusetts) — London (England): The M.I.T. Press, 1966. — xi + 384 p.
- Truesdell C. The Tragicomical History of Thermodynamics, 1822–1854. — New York — Heidelberg — Berlin: Springer-Verlag, 1980. — xii + 372 p. — (Studies in the History of Mathematics and Physical Sciences. Vol. 4). — ISBN 978-1-4613-9446-4.
- Truesdell C., Bharatha S. The Concepts and Logic of Classical Thermodynamics as a Theory of Heat Engines. — New York — Heidelberg — Berlin: Springer-Verlag, 1977. — xvii + 154 p. — ISBN 3-540-07971-8.
- Truesdell C. Rational Thermodynamics. — New York—Berlin—Heidelberg—Tokyo: Springer-Verlag, 1984. — xviii + 578 p. — ISBN 0-387-90874-9.
- Wehrl Alfred. General properties of entropy (англ.) // Reviews of Modern Physics. — American Physical Society, 1978. — Vol. 50, no. 2. — P. 221–260. — doi:10.1103/RevModPhys.50.221.
- Алексеев Г. Н. Энергия и энтропия. — М.: Знание, 1978. — 192 с. — (Жизнь замечательных идей).
- Архаров А. М., Исаев С. И., Кожинов И. А. и др. Теплотехника / Под. общ. ред. В. И. Крутова. — М.: Машиностроение, 1986. — 432 с.
- Ауэрбах Ф. Царица мира и её тень. — 6-е изд. — Одесса: Матезис, 1913. — VIII + 50 с.
- Афанасьева-Эренфест Т. А. Необратимость, односторонность и второе начало термодинамики // Журнал прикладной физики. — 1928. — Vol. 5, № 3–4. — P. 3—30.
- Базаров И. П., Геворкян Э. В., Николаев П. Н. Термодинамика и статистическая физика. Теория равновесных систем. — М.: Издательство Московского университета, 1986. — 311 с.
- Базаров И. П. Термодинамика. — 5-е изд. — СПб.—М.—Краснодар: Лань, 2010. — 384 с. — (Учебники для вузов. Специальная литература). — ISBN 978-5-8114-1003-3.
- Белоконь Н. И. Термодинамика. — М.: Госэнергоиздат, 1954. — 416 с.
- Белоконь Н. И. Основные принципы термодинамики. — М.: Недра, 1968. — 112 с.
- Борн М. Критические замечания по поводу традиционного изложения термодинамики // Развитие современной физики. — М.: Наука, 1964. — С. 223—256.
- Бэр Г. Д. Техническая термодинамика. — М.: Мир, 1977. — 519 с.
- Волькенштейн М. В. Энтропия и информация. — М.: Наука, 1986. — 192 с. — (Проблемы науки и технического прогресса).
- Воронин Г. Ф. Основы термодинамики. — М.: Издательство Моск. университета, 1987. — 192 с.
- Гельфер Я. М. История и методология термодинамики и статистической физики. — 2-е изд., перераб. и доп. — М.: Высшая школа, 1981. — 536 с.
- Герасимов Я. И., Древинг В. П., Ерёмин Е. Н. и др. Курс физической химии / Под общ. ред. Я. И. Герасимова. — 2-е изд. — М.: Химия, 1970. — Т. I. — 592 с.
- Гиббс Дж. В. Термодинамические работы / Пер. с англ. под ред. проф. В. К. Семенченко. — М. — Л.: Гостехтеориздат, 1950. — 492 с. — (Классики естествознания).
- Гиббс Дж. В. Термодинамика. Статистическая механика / Отв. ред. Д. Н. Зубарев. — М.: Наука, 1982. — 584 с. — (Классики науки).
- Глазов В. М. Основы физической химии. — М.: Высшая школа, 1981. — 456 с.
- Гленсдорф П., Пригожин И. Термодинамическая теория структуры, устойчивости и флуктуаций. — М.: Мир, 1973. — 280 с.
- Гухман А. А. Об основаниях термодинамики. — 2-е изд., испр. — М.: Издательство ЛКИ, 2010. — 384 с. — ISBN 978-5-382-01105-9.
- Де Гроот С., Мазур П. Неравновесная термодинамика. — М.: Мир, 1964. — 456 с.
- Жариков В. А. Основы физической геохимии. — М.: Наука; Издательство МГУ, 2005. — 656 с. — (Классический университетский учебник). — ISBN 5-211-04849-0, 5-02-035302-7.
- Жилин П. А. Рациональная механика сплошных сред. — 2-е изд. — СПб.: Издательство Политехн. университета, 2012. — 584 с. — ISBN 978-5-7422-3248-3.
- Жуковский В. С. Термодинамика / Под ред. А. А. Гухмана. — М.: Энергоатомиздат, 1983. — 304 с.
- Залевски К. Феноменологическая и статистическая термодинамика: Краткий курс лекций / Пер. с польск. под. ред. Л. А. Серафимова. — М.: Мир, 1973. — 168 с.
- Зарипов Р. Г. Новые меры и методы в теории информации. — Казань: Издательство Казан. гос. техн. университета, 2005. — 364 с.
- Зоммерфельд А. Термодинамика и статистическая физика / Пер. с нем.. — М.: Издательство иностр. литературы, 1955. — 480 с.
- Игнатович В. Н. Введение в диалектико-материалистическое естествознание. — Киев: Экмо, 2007. — 468 с. — ISBN 978-966-8555-78-7.
- Исаев С. И. Курс химической термодинамики. — 2-е изд., перераб. и доп. — М.: Высшая школа, 1986. — 272 с.
- Квасников И. А. Молекулярная физика. — М.: Эдиториал УРСС, 2009. — 232 с. — ISBN 978-5-901006-37-2.
- Квасников И. А. Термодинамика и статистическая физика. Т. 1: Теория равновесных систем: Термодинамика. — 2-е изд., сущ. перераб. и доп. — М.: Едиториал УРСС, 2002. — 240 с. — ISBN 5-354-00077-7.
- Каратеодори К. Об основах термодинамики (1909) // Развитие современной физики — Отв. ред. Б. Г. Кузнецов. — М.: Наука, 1964. — 331 с. — С. 223—256.
- Кикоин И. К. Рассказы о физике и физиках. — Библиотечка «Квант». Выпуск 53. — М.: Наука, 1986. — 160 с.
- Клаузиус Р. Механическая теория тепла // Второе начало термодинамики. — М.—Л.: Гостехиздат, 1934. — С. 70—158.
- Кричевский И. Р. Понятия и основы термодинамики. — 2-е изд., пересмотр. и доп. — М.: Химия, 1970. — 440 с.
- Кричевский И. Р., Петрянов И. В. Термодинамика для многих. — М.: Педагогика, 1975. — 160 с. — (Библиотечка Детской энциклопедии «Учёные — школьнику»).
- Кубо Р. Термодинамика. — М.: Мир, 1970. — 304 с.
- Ландау Л. Д., Лифшиц Е. М. Статистическая физика. Часть 1. — 5-е изд. — М.: Физматлит, 2002. — 616 с. — (Теоретическая физика в 10 томах. Том 5). — ISBN 5-9221-0054-8.
- Леонова В. Ф. Термодинамика. — М.: Высшая школа, 1968. — 159 с.
- Ляшков В. И. Теоретические основы теплотехники. — М.: Курс; Инфра-М, 2015. — 328 с. — ISBN 978-5-905554-85-8, 978-5-16-0І0639-7.
- Максимов Л. А., Михеенков А. В., Полищук И. Я. Лекции по статистической физике. — Долгопрудный: МФТИ, 2009. — 224 с.
- Морачевский А. Г., Смирнова Н. А., Пиотровская Е. М. и др. Термодинамика равновесия жидкость—пар / Под ред. А. Г. Морачевского. — Л.: Химия, 1989. — 344 с. — ISBN 5-7245-0363-8.
- Мюнстер А. Химическая термодинамика / Пер. с нем. под. ред. чл.-корр. АН СССР Я. И. Герасимова. — М.: Мир, 1971. — 296 с.
- Новиков И. И. Термодинамика. — М.: Машиностроение, 1984. — 592 с.
- Осипов А.И., Уваров А.В. Энтропия и её роль в науке // Сетевой образовательный журнал. — 2004. — Т. 8, № 1. — С. 70—79.
- Петров Н., Бранков Й. Современные проблемы термодинамики. — Пер. с болг. — М.: Мир, 1986. — 287 с.
- Полянин А. Д., Полянин В. Д., Попов В. А. и др. Краткий справочник для инженеров и студентов. — М.: Международная программа образования, 1996. — 432 с. — ISBN 5-7753-0001-7.
- Пригожин И., Кондепуди Д. Современная термодинамика. От тепловых двигателей до диссипативных структур / Пер. с англ. — М.: Мир, 2002. — 462 с.
- Пригожин И., Стенгерс И. Порядок из хаоса: Новый диалог человека с природой. — М.: Прогресс, 1986. — 461 с.
- Путилов К. А. Термодинамика / Отв. ред. М. Х. Карапетьянц. — М.: Наука, 1971. — 376 с.
- Радушкевич Л. В. Курс термодинамики. — М.: Просвещение, 1971. — 288 с.
- Развитие современной физики. Сборник статей / Отв. ред. Кузнецов Б. Г.. — М.: Наука, 1964. — 331 с.
- Свиридов В. В., Свиридов А. В. Физическая химия. — СПб.: Лань, 2016. — 597 с. — ISBN 978-5-8114-2262-3.
- Свиридонов М. Н. Развитие понятия энтропии в работах Т. А. Афанасьевой-Эренфест // История и методология естественных наук. Выпуск X. Физика. — Издательство МГУ, 1971. — P. 112—129.
- Сейдж Б. Х. Термодинамика многокомпонентных систем. — М.: Недра, 1969. — 304 с.
- Семенченко В. К. [www.libgen.io/book/index.php?md5=FB60848D6D70A19DBF1154ECCAE49E7F Избранные главы теоретической физики]. — 2-е изд., испр. и доп. — М.: Просвещение, 1966. — 396 с. (недоступная ссылка)
- Сивухин Д. В. Общий курс физики. Т. II. Термодинамика и молекулярная физика. — 5-е изд., испр. — М.: ФИЗМАТЛИТ, 2005. — 544 с. — ISBN 5-9221-0601-5.
- Сорокин В. С. Макроскопическая необратимость и энтропия. Введение в термодинамику. — М.: ФИЗМАТЛИТ, 2004. — 174 с. — ISBN 5-9221-0507-8.
- Сычёв В. В. Дифференциальные уравнения термодинамики. — 2-е изд., перераб. — М.: Высшая школа, 1991. — 224 с. — ISBN 5-06-002071-1.
- Сычёв В. В. Дифференциальные уравнения термодинамики. — 3-е изд. — М.: Издательство МЭИ, 2010. — 256 с. — ISBN 978-5-383-00584-2.
- Сычёв В. В. Сложные термодинамические системы. — 5-е изд., перераб. и доп.. — М.: Издательский дом МЭИ, 2009. — 296 с. — ISBN 978-5-383-00418-0.
- Тамм М. Е., Третьяков Ю. Д. Неорганическая химия. Том 1. Физико-химические основы неорганической химии / Под. ред. акад. Ю. Д. Третьякова. — М.: Академия, 2004. — 240 с. — (Высшее профессиональное образование). — ISBN 5-7695-1446-9.
- Тер Хаар Д., Вергеланд Г. Элементарная термодинамика / Пер. с англ.. — М.: Мир, 1968. — 220 с..
- Термодинамика необратимых процессов. Лекции в летней международной школе физики им. Энрико Ферми / Под ред. Д. Н. Зубарева. — М.: Издательство иностранной литературы, 1962. — 427 с.
- Трайбус М. Термостатика и термодинамика. — М.: Энергия, 1971. — 503 с.
- Трусделл К. Термодинамика для начинающих // Механика. Периодический сборник переводов иностранных статей. — М.: Мир, 1970. — № 3 (121), с. 116—128.
- Трусделл К. Первоначальный курс рациональной механики сплошных сред / Пер. с англ. под. ред. П. А. Жилина и А. И. Лурье. — М.: Мир, 1975. — 592 с.
- Фен Дж. Машины, энергия, энтропия. — М.: Мир, 1986. — 335 с.
- Fermi, E., Thermodynamics, Prentice Hall (1937). — Русский перевод: Ферми, Энрико, Термодинамика, Харьков: Издательство Харьковского университета, 1969. — 140 с.
- Физическая энциклопедия / Гл. ред. А. М. Прохоров. — М.: Большая Российская энциклопедия, 1998. — Т. 5. — 760 с. — ISBN 5-85270-101-7.
- Хаазе Р. Термодинамика необратимых процессов. — М.: Мир, 1967. — 544 с.
- Черноуцан А. И. Краткий курс физики. — М.: ФИЗМАТЛИТ, 2002. — 320 с. — ISBN 5-9921-0292-3.
- Хазен А. М. Разум природы и разум человека. — М.: РИО «Мособлполиграфиздата»; НТЦ «Университетский», 2000. — 600 с. — ISBN 5-7953-0044-6.
- Хайтун С. Д. [www.libgen.io/book/index.php?md5=BB7A4DF3D04E410AB163CF2D15B4501C Кризис науки как зеркальное отражение кризиса теории познания: Кризис теории познания]. — М.: Ленанд, 2014. — 448 с. — ISBN 978-5-9710-1296-2. (недоступная ссылка)
- Цирлин А. М. Методы оптимизации в необратимой термодинамике и микроэкономике. — М.: Физматлит, 2003. — 416 с.
- Шамбадаль П. Развитие и приложение понятия энтропии / Пер. с франц. — М.: Наука, 1967. — 279 с.
Ссылки[править | править код]
- Термодинамическая энтропия
- Статья «Эволюция как сопротивление энтропии»
|
|
Некоторые внешние ссылки в этой статье ведут на сайты, занесённые в спам-лист. Эти сайты могут нарушать авторские права, быть признаны неавторитетными источниками или по другим причинам быть запрещены в Википедии. Редакторам следует заменить такие ссылки ссылками на соответствующие правилам сайты или библиографическими ссылками на печатные источники либо удалить их (возможно, вместе с подтверждаемым ими содержимым). Список проблемных ссылок
|
Содержание:
- Энтропия
- Принцип существования энтропии
- Принцип возрастания энтропии
- Принцип действия тепловой машины
- Цикл Карно. Максимальный КПД
- Второе начало термодинамики
Энтропия – это физическая величина, используемая для описания термодинамической системы, одна из основных термодинамических величин.
На странице -> решение задач по физике собраны решения задач и заданий с решёнными примерами по всем темам физики.
Энтропия
Энтропия это, в широком смысле, мера хаоса в какой-либо системе. В переводе «энтропия» означает «преобразование».
Энтропия простыми словами — это то, как много информации вам не известно о системе.
В статистической физике энтропия характеризует вероятность осуществления какого-либо макроскопического состояния. Кроме физики, термин широко употребляется в математике: теории информации и математической статистике. В этих областях знания энтропия определяется статистически и называется статистической или информационной энтропией.
В широком смысле, в каком слово часто употребляется в быту, энтропия означает меру сложности, хаотичности или неопределённости системы: чем меньше элементы системы подчинены какому-либо порядку, тем выше энтропия. Величина, противоположная энтропии, именуется негэнтропией или, реже, экстропией.
Принцип существования энтропии
В середине прошлого века было сделано существенное открытие» касающееся обратимых термодинамических процессов. Оказалось, что наряду с внутренней энергией у тела имеется еще одна замечательная функция состояния — энтропия. Также, как и внутренняя энергия, энтропия определяется с точностью до произвольной постоянной. Опыт дает значение разности приращения энтропии. Если тело или система при бесконечно малом переходе из одного состояния в другое при температуре  получает тепло
получает тепло  то отношение
то отношение  является полным дифференциалом некоторой функции
является полным дифференциалом некоторой функции  Эта функция и есть энтропия
Эта функция и есть энтропия
определяющаяся, таким образом, одним из двух эквивалентных равенств:

Утверждение о существовании функции, дифференциал которой есть  носит название принципа существования энтропии и является одним из важнейших законоз природы. Оно является существенной частью второго начала термодинамики, о чем у нас речь пойдет ниже. Открытие этого принципа, как и всего второго начала термодинамики, связано, прежде всего, с именами Карно и Клаузиу-са. Сущность принципа, несмотря на некоторую его абстрактность, легко понять: переход тела из одного состояния во второе может произойти бесчисленным количеством способов (разные кривые на графике, начинающиеся и заканчивающиеся в тех же точках); при этих переходах тело может получать самые различные количества тепла, однако во всех случаях интеграл
носит название принципа существования энтропии и является одним из важнейших законоз природы. Оно является существенной частью второго начала термодинамики, о чем у нас речь пойдет ниже. Открытие этого принципа, как и всего второго начала термодинамики, связано, прежде всего, с именами Карно и Клаузиу-са. Сущность принципа, несмотря на некоторую его абстрактность, легко понять: переход тела из одного состояния во второе может произойти бесчисленным количеством способов (разные кривые на графике, начинающиеся и заканчивающиеся в тех же точках); при этих переходах тело может получать самые различные количества тепла, однако во всех случаях интеграл  будет иметь одинаковые значения. Отношение количества теплоты к той температуре, при которой это тепло было получено,
будет иметь одинаковые значения. Отношение количества теплоты к той температуре, при которой это тепло было получено, называют иногда приведенной теплотой. Так как интеграл всегда можно представить приближенно суммой то изменение энтропии при переходе из одного, состояния в другое равно сумме приведенных теплот. Предположим, что тело, равномерно нагреваясь от 20 до 25 °С, получает при подъеме температуры по одному джоулю тепла на каждый градус. Тогда прирост энтропии будет примерно равен
называют иногда приведенной теплотой. Так как интеграл всегда можно представить приближенно суммой то изменение энтропии при переходе из одного, состояния в другое равно сумме приведенных теплот. Предположим, что тело, равномерно нагреваясь от 20 до 25 °С, получает при подъеме температуры по одному джоулю тепла на каждый градус. Тогда прирост энтропии будет примерно равен

Наиболее просто выражаются изменения энтропии при изотермических процессах:
 где
где  — полученное при процессе тепло. Так, например, при таянии 1 кг льда энтропия вещества возрастает на
— полученное при процессе тепло. Так, например, при таянии 1 кг льда энтропия вещества возрастает на
За нуль энтропии может быть принято значение энтропии любого состояния (кипящей воды, плавящегося льда). Однако в некоторых случаях принимают за нуль значение энтропии вещества при абсолютном нуле температуры. Для этого, впрочем, имеются некоторые теоретические основания (теорема Нернста), на которых мы останавливаться не будем.
Приняв  энтропию вещества при температуре
энтропию вещества при температуре  можно найти по формуле
можно найти по формуле

если нагрев происходит при постоянном давлении. Как видим, чтобы знать энтропию, надо изучить ход теплоемкости с температурой.
Если известно уравнение состояния вещества, то энтропия (с точностью до произвольной постоянной) может быть вычислена весьма просто. По определению Подставляя значение для
Подставляя значение для таким, как его дает первое начало термодинамики, получим
таким, как его дает первое начало термодинамики, получим

При помощи уравнения газового состояния исключим отсюда давление. Получим:  Если взять неопределенный интеграл, то получим выражение энтропии с точностью до произвольной постоянной
Если взять неопределенный интеграл, то получим выражение энтропии с точностью до произвольной постоянной
Можно также взять от  определенный интеграл, пределами которого являются два состояния. Тогда получится выражение для разности энтропий двух состояний
определенный интеграл, пределами которого являются два состояния. Тогда получится выражение для разности энтропий двух состояний
Это — выражения для энтропии идеальных газов. Из формул видно, что энтропия возрастает при повышении температуры, а также при увеличении объема газа. Это, разумеется, полностью совпадает с общим утверждением о повышении энтропии при подводе к телу тепла.
Пример. Покажем, что энтропия действительно есть функция состояния системы. Обратимся к примеру на стр. 151 (рис. 79). Путь 1—2—3. Изменение энтропии

Изменение энтропии

Полное изменение энтропии на пути 1—2—3

Видно, что действительно, каким бы путем ни совершался переход газа из состояния 1 в состояние 3, изменение энтропии одно и то же.
Принцип возрастания энтропии
Как уже говорилось, обратимых процессов, строго говоря, не существует, хотя с точностью, доступной опыту, можно осуществить множество процессов, практически неотличимых от обратимых. Имеются, однако, примеры процессов, которые всегда односторонни и уже поэтому никоим образом не могут быть обращены. Так, например, газ может расшириться сам по себе, но не может сжиматься без приложения внешних сил. Тепло может самопроизвольно переходить от горячего тела к холодному и только при затрате работы (например, электроэнергии) может переходить от холодного тела к более нагретому. При трении кинетическая энергия макроскопического движения всегда превращается во внутреннюю энергию, но никогда не происходит самопроизвольный обратный процесс. Необратимость всех остальных процессов в конечном счете связана с тем, что в каждом из них в той или иной степени присутствует один из перечисленных односторонних процессов. В реальных процессах невозможно избежать ни самопроизвольных расширений, ни трения, ни теплового рассеяния.
Нет ли какого-нибудь общего признака у всех перечисленных односторонних процессов? Оказывается, есть: этот признак состоит в том, что во всех односторонних процессах возрастает энтропия.
В случае теплообмена между двумя телами общее (всей системы) изменение энтропии равно

где  — тепло, полученное более холодным телом,
— тепло, полученное более холодным телом, — тепло, потерянное более горячим телом.
— тепло, потерянное более горячим телом.
Если  больше
больше  так как мы считаем положительным тепло, сообщенное телу. Значит,
так как мы считаем положительным тепло, сообщенное телу. Значит,
т. е. при теплообмене общая энтропия системы, в которой произошел теплообмен, возрастает.
Другой случай. Внутри сосуда с газом происходит интенсивное механическое движение (скажем, вертится колесо). Объем не меняется, температура растет, поэтому энтропия изменится на
Наконец, при расширении в пустоту при неизменной температуре прирост энтропии опять-таки положительный.
опять-таки положительный.
Итак, во всех односторонних процессах энтропия системы возрастает.
Нетрудно понять, какое значение имеет этот вывод для всех необратимых процессов. Так как каждый необратимый процесс сопровождается односторонними явлениями, идущими с повышением энтропии,.то прирост, энтропии у необратимого процесса будет завышен против того прироста, который имел бы место при обратимом переходе. Пусть  — тепло, полученное телом при температуре
— тепло, полученное телом при температуре  в интересующем нас необратимом процессе. Если бы процесс был обратимым, то прирост энтропии равнялся бы
в интересующем нас необратимом процессе. Если бы процесс был обратимым, то прирост энтропии равнялся бы  в реальном процессе прирост энтропии будет больше этой величины:
в реальном процессе прирост энтропии будет больше этой величины:
Если система теплоизолирована, то  и предыдущее утверждение приобретает вид
и предыдущее утверждение приобретает вид
в теплоизолированной системе возможны лишь процессы, идущие с возрастанием энтропии.
Вполне понятно, что энтропия вместе с внутренней энергией являются важнейшими функциями, определяющими термодинамический процесс. Можно сказать, что энтропия является директором-распорядителем процесса, а внутренняя энергия является его бухгалтером: энтропия (определяет направление протекания процесса, энергия «оплачивает расходы» на его проведение.
Если в предыдущие формулы ввести вместо знака  краткой формулой запишется закон энтропии как для обратимых, так и для необратимых процессов:
краткой формулой запишется закон энтропии как для обратимых, так и для необратимых процессов:
Эта формула передает содержание второго начала термодинамики. Для замкнутых систем второе начало говорит: энтропия теплоизолированной системы возрастает или остается неизменной.
Целесообразно объединить оба начала термодинамики одной формулой

удобной для рассмотрения всех практических задач термодинамики.
Принцип возрастания энтропии относится к закрытым системам. Если же система общается со средой, другими словами, если речь идет об открытой системе, то ее энтропия может, разумеется, и убывать.
Ниже будет показано, что процессы молекулярного упорядочения связаны с уменьшением энтропии. Живой организм из неупорядоченной системы малых молекул, получаемых в процессах питания и дыхания, конструирует высокоорганизованные постройки — биологические макромолекулы (стр. 595). При этом энтропия организма падает.
Если представить себе замкнутую систему организм+среда, энтропия которой обязана расти, то ясно, что энтропия среды должна возрастать, перекрывая уменьшение энтропии организма.
Возрастание энтропии среды происходит за счет выделений организма.
Если процесс стационарной, то
Можно сказать, что жизнедеятельность организма состоит в пропускании через себя потока энтропии вещества. При этом энтропия вещества, входящего в организм, меньше энтропии, отдаваемой среде,— организм деградирует продукты питания.
Примеры. 1. В примере на стр. 57 мы рассмотрели неупругое столкновение пули с баллистическим маятником и выяснили, что при ударе в системе пуля — маятник исчезает 3920 Дж механической энергии. Это значит, что  было необратимым образом передано маятнику от пули посредством теплопроводности. Если предположить, что процесс был изотермическим (т. е. теплопроводность маятника чрезвычайно велика) и температура системы, скажем,
было необратимым образом передано маятнику от пули посредством теплопроводности. Если предположить, что процесс был изотермическим (т. е. теплопроводность маятника чрезвычайно велика) и температура системы, скажем,  то в этом необратимом процессе энтропия системы возросла на
то в этом необратимом процессе энтропия системы возросла на

2. Детский резиновый мяч массой 0,3 кг после падения с высоты 2 м подпрыгивает на I м от пола. В этом изотермическом процессе (пусть необратимо передается
необратимо передается  т. е. энтропия системы мяч — пол возросла на
т. е. энтропия системы мяч — пол возросла на
Если бы мяч и пол были абсолютно упругими, то энтропия не менялась бы  движение мяча продолжалось бы вечно.
движение мяча продолжалось бы вечно.
3. Рассмотрим необратимый процесс передачи тепла от парового котла к конденсатору. Пусть паровой котел находится при температуре  а конденсатор — при
а конденсатор — при  При тепловой мощности котла 10 000 кВт и к. п. д. 25% ежесекундно от котла к конденсатору будет переноситься
При тепловой мощности котла 10 000 кВт и к. п. д. 25% ежесекундно от котла к конденсатору будет переноситься  Для котла, теряющего теплоту, это
Для котла, теряющего теплоту, это  будет отрицательным, т. е. его энтропия убывает; у конденсатора энтропия растет. Но так как
будет отрицательным, т. е. его энтропия убывает; у конденсатора энтропия растет. Но так как  то энтропия системы котел — конденсатор за каждую секунду возрастает на
то энтропия системы котел — конденсатор за каждую секунду возрастает на
Принцип действия тепловой машины
Тепловая машина превращает тепло в работу, иначе говоря, забирает тепло от одних тел и передает его другим телам в форме механической работы. Для того чтобы осуществить это превращение, надо располагать двумя различно нагретыми телами, между которыми возможен теплообмен. Для краткости будем называть более горячее тело нагревателем, а более холодное — холодильником. При наличии таких двух тел процесс превращения тепла в работу рисуется следующим образом: способное расшириться тело (рабочее тело) приводится в контакт с нагревателем. Тепло ( отбирается от нагревателя и затрачивается на работу расширения
( отбирается от нагревателя и затрачивается на работу расширения  которая отдается окружающим телам. Далее, рабочее тело приводится в контакт с холодильником, которому оно отдает тепло
которая отдается окружающим телам. Далее, рабочее тело приводится в контакт с холодильником, которому оно отдает тепло  за счет работы
за счет работы  совершаемой внешними силами над рабочим телом.
совершаемой внешними силами над рабочим телом.
Чтобы получить непрерывно действующую тепловую машину, необходимо закончить такт сжатия в той точке, в которой начался такт расширения; короче, процесс должен быть циклическим. Рабочее тело по проведении каждого цикла возвращается в исходное состояние. Закон сохранения энергии требует поэтому, чтобы энергия, полученная от окружающих тел, равнялась энергии, переданной окружающим телам. От среды получено: тепло при расширении и работа
при расширении и работа  при сжатии рабочего тела. Среде отдано: работа
при сжатии рабочего тела. Среде отдано: работа  при расширении тела
при расширении тела  и тепло при сжатии. Следовательно,
и тепло при сжатии. Следовательно, При проведении цикла по часовой стрелке работа сжатия меньше работы расширения. Поэтому последнее равенство выражает тот простой факт, что чистая работа, переданная рабочим телом внешней среде, равна разности теплот, полученной от нагревателя и отданной холодильнику. Соответственно коэффициент полезного действия цикла, а значит, и всей машины, будет равен
При проведении цикла по часовой стрелке работа сжатия меньше работы расширения. Поэтому последнее равенство выражает тот простой факт, что чистая работа, переданная рабочим телом внешней среде, равна разности теплот, полученной от нагревателя и отданной холодильнику. Соответственно коэффициент полезного действия цикла, а значит, и всей машины, будет равен 
Описанный процесс действия тепловой машины является, разумеется, абстрактной схемой. Однако наиболее существенные черты каждого теплового двигателя передаются этой схемой. Рабочим телом является расширяющийся и сжимающийся газ или пар, роль холодильника играет окружающая среда. Нагревателем служит паровой котел или, в двигателях внутреннего сгорания, горючая смесь.
Те же три системы являются необходимыми и для холодильной машины, в которой цикл протекает в обратную сторону. Принцип работы этой машины заключается в следующем: расширение рабочего тела производится тогда, когда оно находится в контакте с холодильником. Этим холодное тело охлаждается еще больше, что и является задачей холодильной машины. Далее, чтобы цикл стал возможным, нужно произвести сжатие рабочего тела и передать тепло, полученное от холодильника. Это выполняется при контакте рабочего тела с нагревателем. Таким образом, более горячее тело нагревается еще больше. «Противоестественный» переход тепла от тела менее нагретого к телу более нагретому «оплачивается» работой. Действительно, при совершении цикла против часовой стрелки равенство энергии, переданной среде, и энергии, отнятой от среды  где мы по-прежнему
где мы по-прежнему
индекс 1 относим к части процесса, протекающей при контакте с более горячим телом), имеет следующий смысл: количество тепла, отнятое от системы, должно быть скомпенсировано равным количеством механической работы.
Второе начало термодинамики накладывает некоторое условие на действие тепловой машины. Если предполагать процесс обратимым, то изменение энтропии рабочего тела после прохождения цикла должно равняться нулю. Можно сказать и иначе: изменение энтропии в процессе расширения должно равняться (с обратным знаком) изменению энтропии при сжатии, т. е.

В случае же необратимого процесса энтропия замкнутой системы, состоящей из нагревателя, холодильника и рабочего тела, возрастет и поэтому

(Напоминаем, что  есть алгебраическая величина. Тепло, поступившее в систему, считается положительным.) Подсчитывая значения этих интегралов для конкретных процессов, можно в ряде случаев довольно просто найти значение максимального коэффициента полезного действия того или иного цикла тепловой машины.
есть алгебраическая величина. Тепло, поступившее в систему, считается положительным.) Подсчитывая значения этих интегралов для конкретных процессов, можно в ряде случаев довольно просто найти значение максимального коэффициента полезного действия того или иного цикла тепловой машины.
Цикл Карно. Максимальный КПД
Сейчас мы задаемся целью найти выражение предельно большого коэффициента полезного действия тепловой машины, достижимого для идеальной машины, работающей без потерь на обратимом цикле.
Прежде всего рассмотрим теоретический четырехтактный цикл Карно, изображенный на рис. 81. Цикл Карно состоит из двух изотерм (для температур  и
и  и двух адиабат. Первый такт процесса пусть будет изотермическое расширение от состояния 1 к состоянию 2 — рабочее тело находится в контакте с нагревателем, имеющим температуру
и двух адиабат. Первый такт процесса пусть будет изотермическое расширение от состояния 1 к состоянию 2 — рабочее тело находится в контакте с нагревателем, имеющим температуру  и процесс проводится весьма медленно. По достижении состояния 2 контакт с нагревателем нарушается, тело теплоизолируется и ему предоставляется возможность дополнительно расшириться.
и процесс проводится весьма медленно. По достижении состояния 2 контакт с нагревателем нарушается, тело теплоизолируется и ему предоставляется возможность дополнительно расшириться.

Работа происходит за счет внутренней энергии и температура тела пусть падает до  Начиная с этой точки (состояние 3) начинается двухтактное сжатие. Тело сообщается с холодильником при температуре
Начиная с этой точки (состояние 3) начинается двухтактное сжатие. Тело сообщается с холодильником при температуре  и изотермически сжимается до состояния 4. Здесь опять тело теплоизолируется и сжатие продолжается уже адиабатическим путем с нагреванием рабочего тела за счет совершаемой работы до начальной температуры
и изотермически сжимается до состояния 4. Здесь опять тело теплоизолируется и сжатие продолжается уже адиабатическим путем с нагреванием рабочего тела за счет совершаемой работы до начальной температуры 
Адиабатические процессы в цикле Карно носят вспомогательный характер: они помогают перейти с одной изотермы на другую. В энергетическом балансе эти процессы не участвуют, так как работа адиабатического расширения  и работа сжатия
и работа сжатия  компенсируют друг друга.
компенсируют друг друга.
В адиабатическом процессе энтропия системы не меняется. При изотермическом расширении энтропия нагревателя уменьшается на величину  энтропия холодильника возрастает на
энтропия холодильника возрастает на  Энтропия рабочего тела, вернувшегося в исходное состояние, остается неизменной. Если процесс обратим, то
Энтропия рабочего тела, вернувшегося в исходное состояние, остается неизменной. Если процесс обратим, то  В необратимых процессах энтропия всей системы, состоящей из холодильника, нагревателя и рабочего тела, возрастает и прирост энтропии
В необратимых процессах энтропия всей системы, состоящей из холодильника, нагревателя и рабочего тела, возрастает и прирост энтропии  больше убыли
больше убыли


откуда и следовательно, максимальный коэффициент полезного действия цикла Карно равен
и следовательно, максимальный коэффициент полезного действия цикла Карно равен


К- п. д. цикла определяется температурами холодильника и нагревателя. Чем больше перепад температуры, тем выше к. п. д. машины. Нетрудно видеть, что коэффициент полезного действия цикла Карно дает оптимальное значение к. п. д. Нет лучшего цикла, чем цикл Карно, и в этом смысле он должен являться образцом для конструкторов тепловых машин, они должны стремиться как можно более приблизить реальные циклы к циклу этой идеальной машины.
Доказательство не составит труда. На рис. 82 показан произвольный цикл, вписанный в цикл
Карно. Уменьшение энтропии нагревателя может быть представлено интегралом
 для которого несомненно справедливо неравенстве
для которого несомненно справедливо неравенстве
так как  — самое большое число из тех значений, которые пробегает
— самое большое число из тех значений, которые пробегает  при интегрировании. Увеличение энтропии холодильника выразится интегралом
при интегрировании. Увеличение энтропии холодильника выразится интегралом для которого справедливо неравенство
для которого справедливо неравенство  так как
так как  — самое маленькое число из тех значений, которые пробегает
— самое маленькое число из тех значений, которые пробегает  при интегрировании. При обратимом процессе
при интегрировании. При обратимом процессе
следовательно, что и дает условие
что и дает условие
Итак, из всех возможных циклических процессов максимальным к. п. д. обладает цикл Карно.
Формула максимального к. п. д. показывает причину низкого к. п. д. паровых машин. При  к. п. д. равен 25%. Однако ведь это — максимальный коэффициент полезного действия, он достижим для идеальной машины, работающей обратимо без каких бы то ни было потерь энергии. Не приходится удивляться, что в реальных паровых машинах к. п. д. ниже 10%. В курсе теплотехники рассказывается о путях, которыми идет техника для увеличения коэффициента полезного действия. Ясно, что наиболее существенным является повышение температуры нагревателя, т. е. пара или горючей смеси.
к. п. д. равен 25%. Однако ведь это — максимальный коэффициент полезного действия, он достижим для идеальной машины, работающей обратимо без каких бы то ни было потерь энергии. Не приходится удивляться, что в реальных паровых машинах к. п. д. ниже 10%. В курсе теплотехники рассказывается о путях, которыми идет техника для увеличения коэффициента полезного действия. Ясно, что наиболее существенным является повышение температуры нагревателя, т. е. пара или горючей смеси.
Второе начало термодинамики
Как было указано выше, второе начало термодинамики состоит в утверждении, что энтропия в теплоизолированной системе возрастает. Это утверждение может показаться несколько абстрактным. Кроме того, приведенная формулировка не соответствует историческому развитию идей. Имея в виду огромную, значимость этого закона природы, надо кратко остановиться на других существующих формулировках второго начала термодинамики и показать их эквивалентность приведенной выше.
Исторически второе начало термодинамики вошло в науку в виде постулата Томсона о невозможности создания вечного двигателя второго рода. Вечным двигателем первого рода называют машину, создающую работу «из ничего», т. е. машину, работа которой нарушает первое начало термодинамики. Вечным двигателем второго рода называют такой двигатель, который производит работу при помощи периодически действующей машины за счет одного лишь отнятия теплоты от окружающей среды. Такой двигатель, будь он возможен, был бы практически вечным, так как запас энергии в окружающей среде почти безграничен и охлаждение, скажем, воды океанов на один градус дало бы непредставимо огромную энергию. Масса воды в мировом океане по порядку величины составляет  При охлаждении всей этой массы воды лишь на
При охлаждении всей этой массы воды лишь на  выделилось бы
выделилось бы  тепла, что эквивалентно полному сжиганию
тепла, что эквивалентно полному сжиганию  угля. Железнодорожный состав, нагруженный этим количеством угля, растянулся бы на расстояние
угля. Железнодорожный состав, нагруженный этим количеством угля, растянулся бы на расстояние  что по порядку величины совпадает с размерами солнечной системы!
что по порядку величины совпадает с размерами солнечной системы!
Вечный двигатель второго рода — это тепловая машина, работающая с нагревателем, но без холодильника. Такая машина могла бы поработать один такт — газ, находящийся в сосуде с поршнем, мог бы расшириться, но на этом работа двигателя и закончилась бы, так как для продолжения действия машины тепло, полученное газом, необходимо передать холодильнику. Формально невозможность вечного двигателя второго рода видна из формулы максимального к. п. д. При отсутствии теплового перепада  максимальное значение к. п. д. равно нулю.
максимальное значение к. п. д. равно нулю.
Невозможно осуществить периодически действующий вечный двигатель, комбинируя изотермическое расширение с адиабатическим процессом сжатия. Такой процесс невозможен, даже если бы удалось его сделать обратимым. При изотермическом расширении рабочего тела энтропия падает. Значит, процесс сжатия должен приводить к возрастанию энтропии. Этого, однако, не может сделать адиабатический процесс, так как он проходит при постоянной энтропии.
Вполне соответствует принятой здесь формулировке второго начала термодинамики также постулат Клаузиуса, который состоит в утверждении о невозможности перехода тепла от менее нагретого тела к более нагретому без компенсации. Процесс, противоречащий постулату Клаузиуса, протекает с уменьшением энтропии; это свойство энтропии было показано с самого начала.
Услуги по физике:
- Заказать физику
- Заказать контрольную работу по физике
- Помощь по физике
Лекции по физике:
- Физические величины и их измерение
- Основные законы механики
- Прямолинейное равномерное движение
- Прямолинейное равнопеременное движение
- Сила
- Масса
- Взаимодействия тел
- Механическая энергия
- Импульс
- Вращение твердого тела
- Криволинейное движение тел
- Колебания
- Колебания и волны
- Механические колебания и волны
- Бегущая волна
- Стоячие волны
- Акустика
- Звук
- Звук и ультразвук
- Движение жидкости и газа
- Молекулярно-кинетическая теория
- Молекулярно-кинетическая теория строения вещества
- Молекулярно – кинетическая теория газообразного состояния вещества
- Теплота и работа
- Температура и теплота
- Термодинамические процессы
- Идеальный газ
- Уравнение состояния идеального газа
- Изменение внутренней энергии
- Переход вещества из жидкого состояния в газообразное и обратно
- Кипение, свойства паров, критическое состояние вещества
- Водяной пар в атмосфере
- Плавление и кристаллизация
- Тепловое расширение тел
- Процессы перехода из одного агрегатного состояния в другое
- Тепловое расширение твердых и жидких тел
- Свойства газов
- Свойства жидкостей
- Свойства твёрдых тел
- Изменение агрегатного состояния вещества
- Тепловые двигатели
- Электрическое поле
- Постоянный ток
- Переменный ток
- Магнитное поле
- Электромагнитное поле
- Электромагнитное излучение
- Электрический заряд (Закон Кулона)
- Электрический ток в металлах
- Электрический ток в электролитах
- Электрический ток в газах и в вакууме
- Электрический ток в полупроводниках
- Электромагнитная индукция
- Работа, мощность и тепловое действие электрического тока
- Термоэлектрические явления
- Распространение электромагнитных волн
- Интерференционные явления
- Рассеяние
- Дифракция рентгеновских лучей на кристалле
- Двойное лучепреломление
- Магнитное поле и электромагнитная индукция
- Электромагнитные колебания и волны
- Природа света
- Распространение света
- Отражение и преломление света
- Оптические приборы и зрение
- Волновые свойства света
- Действия света
- Линзы и получение изображений с помощью линз
- Оптические приборы и глаз
- Фотометрия
- Излучение и спектры
- Квантовые свойства излучения
- Специальная теория относительности в физике
- Теория относительности
- Квантовая теория и природа поля
- Строение и свойства вещества
- Физика атомного ядра
- Строение атома
2.2.1. Расчёт энтропии для равновесных (обратимых) процессов.
Второй
закон термодинамики в виде
![]() ,
,
записанный для равновесных процессов,
позволяет вычислить не абсолютное
значение энтропии, а только разность
энтропий в двух состояниях системы.
![]() . (2.4)
. (2.4)
Рассмотрим
для 1 моля вещества:
а)
Изотермические процессы
(T
= const).
При
постоянной температуре протекают
процессы фазовых превращений веществ:
плавление, испарение и другие. При
равновесном протекании этих процессов
давление сохраняется обычно постоянным,
поэтому
![]() и
и
![]() , (2.5)
, (2.5)
где
![]() – энтальпия фазового перехода.
– энтальпия фазового перехода.
б)
Изобарные процессы
(р
= const).
Если
нагревание происходит при постоянном
давлении, то
![]() , (2.6)
, (2.6)
где
n
– число молей вещества. Тогда
 . (2.7)
. (2.7)
Пример
2.1. Определить
изменение энтропии при нагреве 1 моль
Al от 25 до 6000С,
если для него в этом интервале теплоёмкость
зависит от температуры следующим
образом:
![]()
![]() ,
,
(Дж/моль К).
Решение.
Согласно
уравнению (2.7) имеем:
 ,
,
![]() (Дж/моль К).
(Дж/моль К).
с)
Изохорные процессы
(V
= const).
Если
нагревание происходит при постоянном
объёме, то
![]() . (2.8)
. (2.8)
Тогда
 . (2.9)
. (2.9)
Для 1 моля
идеального газа справедливо:
а)
При изменении объёма и температуры
![]() , (2.10)
, (2.10)
с
учетом, что
![]() .
.
б)
При изменении давления и температуры
![]() . (2.11)
. (2.11)
Для
любого вещества при любой температуре
можно определить и абсолютное значение
энтропии, если воспользоваться постулатом
Планка:
энтропия
правильно образованного кристалла
любого индивидуального вещества при
абсолютном нуле равна нулю.
Если
вещество при температуре Т
находится в газообразном состоянии, то
его абсолютная энтропия может быть
вычислена по формуле:
 . (2.12)
. (2.12)
2.2.2. Расчёт изменения энтропии в ходе химической реакции.
Расчёт
изменения энтропии в ходе химической
реакции проводится по формуле:
![]() , (2.13)
, (2.13)
где
![]() – стандартные энтропии веществ приТ
– стандартные энтропии веществ приТ
= 298,15 К.
Каждое
вещество характеризуется стандартной
энтропией
![]() – энтропией 1 моль вещества при 298.15 К
– энтропией 1 моль вещества при 298.15 К
и давлении 1 атм. Значения энтропии
имеют размерность Дж/(моль К) или кал/(моль
К).Стандартные
энтропии простых веществ не равны нулю.
2.2.3. Расчёт изменения энтропии в ходе самопроизвольных (необратимых) процессов.
Для
необратимых процессов
![]() и уравнение (2.4) не применимо. Энтропия
и уравнение (2.4) не применимо. Энтропия
– функция состояния и её изменение не
зависит от пути процесса, а определяется
конечным и начальным состоянием системы.
Изменение энтропии в любом неравновесном
процессе можно вычислить, заменяя его
некоторой совокупностью равновесных
процессов, просходящими между теми же
начальными и конечными состояниями,
для каждого из которых можно рассчитать
значение![]() .
.
Тогда:
![]() . (2.14)
. (2.14)
2.3. Энергия гиббса, энергия гельмгольца. Уравнение гиббса–гельмгольца.
В изолированных
системах энтропия только увеличивается
и при равновесии достигает максимума.
Поэтому она может быть использована в
качестве критерия протекания
самопроизвольных процессов в таких
системах. Однако на практике большинство
процессов происходит в неизолированных
системах, вследствие чего для них надо
выбрать свои критерии направления
самопроизвольных процессов и достижения
равновесия в этих системах. Такими
критериями являются другие термодинамические
функции, отличные от энтропии и внутренней
энергии. Они подобраны таким образом,
что с их помощью можно определить в
явной форме все термодинамические
параметры изучаемой системы. Все они
являются функциями состояния и при
переходе системы из одного положения
в другое меняются однозначно. При
достижении системой равновесного
состояния каждая из функций проходит
через минимальное значение. Такие
свойства обуславливают широкое применение
этих функций при аналитическом методе
решения различных задач термодинамических
исследований.
Следует
отметить, что такие функции часто
называют характеристическими.
Характеристической
функцией
называется такая функция состояния
системы, посредством которой и её
производных могут быть выражены в явной
форме все термодинамические свойства
системы.
Согласно
первому закону термодинамики:
A
= Q
– dU. (2.15)
Подставив
сюда извесное соотношение Q
≤ TdS, получим
A
≤ TdS
– dU, (2.16)
где
знак равенства относится к обратимым
равновесным процессам, а знак неравенства
— к необратимым. Проинтегрируем
(2.16) при
Т
= const:
AT
≤
T(S2
– S1)
– (U2
– U1)
=
(U1
– TS1)
– (U2
– TS2). (2.17)
Функция (U
– TS)
играет большую роль при изучении
равновесия в изотермических процессах.
Её называют изохорно-изотермическим
потенциалом
или энергией
Гельмгольца
и обозначают символом F.
При этом для всякого изотермического
процесса:
dF
= dU –
TdS, (2.18)
∆F
= ∆U –
T∆S, (2.19)
а
максимальная работа в изотермическом
процессе
(AТ)max
= –∆F. (2.20)
Функция
F определяет направление и предел течения
самопроизвольных процессов, протекающих
при постоянных температуре и объёме.
Близкой
к изохорно-изотермическому потенциалу
является функция, определяющая направление
и предел самопроизвольного протекания
процессов для систем, находящихся при
постоянных температуре и давлении. Эта
функция называется изобарно-изотер-мическим
потенциалом
или энергией
Гиббса,
обозначается символом G
и определяется как
G
= H – TS. (2.21)
или
G
= U – TS
+ pV = F
+ pV. (2.22)
Пусть р
= const, тогда
AT
≤ –∆F = F1
– F2, (2.23)
AT
+ p(V2
– V1)
≤
F1
– F2, (2.24)
AT
≤ (F1+pV1)
– (F2
+ pV2)
=
G1
– G2, (2.25)
где
AT
– полезная работа (любая работа кроме
работы расширения). Тогда
AT ≤
–∆G. (2.26)
При
этом для изотермических процессов
![]() , (2.27)
, (2.27)
![]() , (2.28)
, (2.28)
и
максимальная работа в изотермическом
процессе
![]() , (2.29)
, (2.29)
где
![]() ,
,
т.е.
максимальная полезная работа равна
максимальной работе изотермического
процесса за вычетом работы против сил
внешнего давления. Функции G
и F
называются термодинамическими
потенциалами,
потому что в определённых условиях
стремятся к минимуму при протекании
самопроизвольных процессов.
Пусть
![]() ,
,
тогда
![]() . (2.30)
. (2.30)
1). Система
при T, V = const
![]() ,
,
т. е. ΔF ≤ 0.
Условие равновесия в изохорно-изотермической
системе: dF = 0,
ΔF = 0,
F
= Fmin.
2). Система
при р, T = const.
Тогда
![]() Условие равновесия в изобарно-изотерми-ческой
Условие равновесия в изобарно-изотерми-ческой
системе:dG = 0,
ΔG = 0,
G
= Gmin.
Вывод:
в системах, находящихся при постоянных
температуре и объёме, самопроизвольно
могут протекать только те процессы,
которые сопровождаются уменьшением
энергии Гельмгольца F,
причем пределом их протекания, т.е.
условием равновесия, является достижение
некоторого минимального для данных
условий значения функции F;
в системах же, находящихся при постоянных
температуре и давлении, самопроизвольно
могут протекать только те процессы,
которые сопровождаются уменьшением
энергии Гиббса G,
причём пределом их протекания, т.е.
условием равновесия, служит достижение
некоторого минимального для данных
условий значения функции G.
Получим соотношения,
которые описывают зависимость
![]() и
и![]() от температуры. В общем случае (и для
от температуры. В общем случае (и для
химических реакций):
![]() , (2.31)
, (2.31)
![]() . (2.32)
. (2.32)
Функция
состояния обладает свойствами полного
дифференциала, т.е. если
![]() ,
,
то
![]() . (2.33)
. (2.33)
С
другой стороны:
F
= U – TS, (2.34)
dF
= dU – TdS
– SdT. (2.35)
С
учетом того, что
dU
=
![]() , (2.36)
, (2.36)
получаем
![]() . (2.37)
. (2.37)
При
сравнении уравнений (2.37) и (2.33) видно,
что
![]()
![]() , (2.38)
, (2.38)
![]() . (2.39)
. (2.39)
Аналогично для
![]() ,
,
получаем:
![]() , (2.40)
, (2.40)
![]()
![]() , (2.41)
, (2.41)
![]() . (2.42)
. (2.42)
Подставляя
соотношения (2.39) и (2.42) в уравнения (2.31)
и (2.32), соответственно, получаем:
![]() , (2.43)
, (2.43)
![]() . (2.44)
. (2.44)
Последние
два равенства и есть искомые зависимости
![]() и
и![]() от температуры и их называютуравнениями
от температуры и их называютуравнениями
Гиббса-Гельмгольца.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Энтропия? Это просто!
Время на прочтение
7 мин
Количество просмотров 406K
Этот пост является вольным переводом ответа, который Mark Eichenlaub дал на вопрос What’s an intuitive way to understand entropy?, заданный на сайте Quora
Энтропия. Пожалуй, это одно из самых сложных для понимания понятий, с которым вы можете встретиться в курсе физики, по крайней мере если говорить о физике классической. Мало кто из выпускников физических факультетов может объяснить, что это такое. Большинство проблем с пониманием энтропии, однако, можно снять, если понять одну вещь. Энтропия качественно отличается от других термодинамических величин: таких как давление, объём или внутренняя энергия, потому что является свойством не системы, а того, как мы эту систему рассматриваем. К сожалению в курсе термодинамики её обычно рассматривают наравне с другими термодинамическими функциями, что усугубляет непонимание.
Так что же такое энтропия?
Если в двух словах, то
Энтропия — это то, как много информации вам не известно о системе
Например, если вы спросите меня, где я живу, и я отвечу: в России, то моя энтропия для вас будет высока, всё-таки Россия большая страна. Если же я назову вам свой почтовый индекс: 603081, то моя энтропия для вас понизится, поскольку вы получите больше информации.
Почтовый индекс содержит шесть цифр, то есть я дал вам шесть символов информации. Энтропия вашего знания обо мне понизилась приблизительно на 6 символов. (На самом деле, не совсем, потому что некоторые индексы отвечают большему количеству адресов, а некоторые — меньшему, но мы этим пренебрежём).

Или рассмотрим другой пример. Пусть у меня есть десять игральных костей (шестигранных), и выбросив их, я вам сообщаю, что их сумма равна 30. Зная только это, вы не можете сказать, какие конкретно цифры на каждой из костей — вам не хватает информации. Эти конкретные цифры на костях в статистической физике называют микросостояниями, а общую сумму (30 в нашем случае) — макросостоянием. Существует 2 930 455 микросостояний, которые отвечают сумме равной 30. Так что энтропия этого макросостояния равна приблизительно 6,5 символам (половинка появляется из-за того, что при нумерации микросостояний по порядку в седьмом разряде вам доступны не все цифры, а только 0, 1 и 2).
А что если бы я вам сказал, что сумма равна 59? Для этого макросостояния существует всего 10 возможных микросостояний, так что его энтропия равна всего лишь одному символу. Как видите, разные макросостояния имеют разные энтропии.
Пусть теперь я вам скажу, что сумма первых пяти костей 13, а сумма остальных пяти — 17, так что общая сумма снова 30. У вас, однако, в этом случае имеется больше информации, поэтому энтропия системы для вас должна упасть. И, действительно, 13 на пяти костях можно получить 420-ю разными способами, а 17 — 780-ю, то есть полное число микросостояний составит всего лишь 420х780 = 327 600. Энтропия такой системы приблизительно на один символ меньше, чем в первом примере.
Мы измеряем энтропию как количество символов, необходимых для записи числа микросостояний. Математически это количество определяется как логарифм, поэтому обозначив энтропию символом S, а число микросостояний символом Ω, мы можем записать:
S = log Ω
Это есть ничто иное как формула Больцмана (с точностью до множителя k, который зависит от выбранных единиц измерения) для энтропии. Если макросостоянию отвечают одно микросостояние, его энтропия по этой формуле равна нулю. Если у вас есть две системы, то полная энтропия равна сумме энтропий каждой из этих систем, потому что log(AB) = log A + log B.

Из приведённого выше описания становится понятно, почему не следует думать об энтропии как о собственном свойстве системы. У системы есть опеделённые внутренняя энергия, импульс, заряд, но у неё нет определённой энтропии: энтропия десяти костей зависит от того, известна вам только их полная сумма, или также и частные суммы пятёрок костей.
Другими словами, энтропия — это то, как мы описываем систему. И это делает её сильно отличной от других величин, с которыми принято работать в физике.
Физический пример: газ под поршнем
Классической системой, которую рассматривают в физике, является газ, находящийся в сосуде под поршнем. Микросостояние газа — это положение и импульс (скорость) каждой его молекулы. Это эквивалентно тому, что вы знаете значение, выпавшее на каждой кости в рассмотренном раньше примере. Макросостояние газа описывается такими величинами как давление, плотность, объём, химический состав. Это как сумма значений, выпавших на костях.
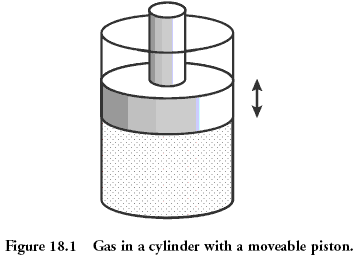
Величины, описывающие макросостояние, могут быть связаны друг с другом через так называемое «уравнение состояния». Именно наличие этой связи позволяет, не зная микросостояний, предсказывать, что будет с нашей системой, если начать её нагревать или перемещать поршень. Для идеального газа уравнение состояния имеет простой вид:
p = ρT
хотя вы, скорее всего, лучше знакомы с уравнением Клапейрона — Менделеева pV = νRT — это то же самое уравнение, только с добавлением пары констант, чтобы вас запутать. Чем больше микросостояний отвечают данному макросостоянию, то есть чем больше частиц входят в состав нашей системы, тем лучше уравнение состояния её описывают. Для газа характерные значения числа частиц равны числу Авогадро, то есть составляют порядка 1023.
Величины типа давления, температуры и плотности называются усреднёнными, поскольку являются усреднённым проявлением постоянно сменяющих друг друга микросостояний, отвечающих данному макросостоянию (или, вернее, близким к нему макросостояниям). Чтобы узнать в каком микросостоянии находится система, нам надо очень много информации — мы должны знать положение и скорость каждой частицы. Количество этой информации и называется энтропией.
Как меняется энтропия с изменением макросостояния? Это легко понять. Например, если мы немного нагреем газ, то скорость его частиц возрастёт, следовательно, возрастёт и степень нашего незнания об этой скорости, то есть энтропия вырастет. Или, если мы увеличим объём газа, отведя поршень, увеличится степень нашего незнания положения частиц, и энтропия также вырастет.
Твёрдые тела и потенциальная энергия
Если мы рассмотрим вместо газа какое-нибудь твёрдое тело, особенно с упорядоченной структурой, как в кристаллах, например, кусок металла, то его энтропия будет невелика. Почему? Потому что зная положение одного атома в такой структуре, вы знаете и положение всех остальных (они же выстроены в правильную кристаллическую структуру), скорости же атомов невелики, потому что они не могут улететь далеко от своего положения и лишь немного колеблются вокруг положения равновесия.
Если кусок металла находится в поле тяготения (например, поднят над поверхностью Земли), то потенциальная энергия каждого атома в металле приблизительно равна потенциальной энергии других атомов, и связанная с этой энергией энтропия низка. Это отличает потенциальную энергию от кинетической, которая для теплового движения может сильно меняться от атома к атому.
Если кусок металла, поднятый на некоторую высоту, отпустить, то его потенциальная энергия будет переходить в кинетическую энергию, но энтропия возрастать практически не будет, потому что все атомы будут двигаться приблизительно одинаково. Но когда кусок упадёт на землю, во время удара атомы металла получат случайное направление движения, и энтропия резко увеличится. Кинетическая энергия направленного движения перейдёт в кинетическую энергию теплового движения. Перед ударом мы приблизительно знали, как движется каждый атом, теперь мы эту информацию потеряли.
Понимаем второй закон термодинамики
Второй закон термодинамики утверждает, что энтропия (замкнутой системы) никогда не уменьшается. Мы теперь можем понять, почему: потому что невозможно внезапно получить больше информации о микросостояниях. Как только вы потеряли некую информацию о микросостоянии (как во время удара куска металла об землю), вы не можете вернуть её назад.

Давайте вернёмся обратно к игральным костям. Вспомним, что макросостояние с суммой 59 имеет очень низкую энтропию, но и получить его не так-то просто. Если бросать кости раз за разом, то будут выпадать те суммы (макросостояния), которым отвечает большее количество микросостояний, то есть будут реализовываться макросостояния с большой энтропией. Самой большой энтропией обладает сумма 35, и именно она и будет выпадать чаще других. Именно об этом и говорит второй закон термодинамики. Любое случайное (неконтролируемое) взаимодействие приводит к росту энтропии, по крайней мере до тех пор, пока она не достигнет своего максимума.
Перемешивание газов
И ещё один пример, чтобы закрепить сказанное. Пусть у нас имеется контейнер, в котором находятся два газа, разделённых расположенной посередине контейнера перегородкой. Назовём молекулы одного газа синими, а другого — красными.
Если открыть перегородку, газы начнут перемешиваться, потому что число микросостояний, в которых газы перемешаны, намного больше, чем микросостояний, в которых они разделены, и все микросостояния, естественно, равновероятны. Когда мы открыли перегородку, для каждой молекулы мы потеряли информацию о том, с какой стороны перегородки она теперь находится. Если молекул было N, то утеряно N бит информации (биты и символы, в данном контексте, это, фактически, одно и тоже, и отличаются только неким постоянным множителем).
Разбираемся с демоном Максвелла
Ну и напоследок рассмотрим решение в рамках нашей парадигмы знаменитого парадокса демона Максвелла. Напомню, что он заключается в следующем. Пусть у нас есть перемешанные газы из синих и красных молекул. Поставим обратно перегородку, проделав в ней небольшое отверстие, в которое посадим воображаемого демона. Его задача — пропускать слева направо только красных, и справа налево только синих. Очевидно, что через некоторое время газы снова будут разделены: все синие молекулы окажутся слева от перегородки, а все красные — справа.
Получается, что наш демон понизил энтропию системы. С демоном ничего не случилось, то есть его энтропия не изменилась, а система у нас была закрытой. Получается, что мы нашли пример, когда второй закон термодинамики не выполняется! Как такое оказалось возможно?
Решается этот парадокс, однако, очень просто. Ведь энтропия — это свойство не системы, а нашего знания об этой системе. Мы с вами знаем о системе мало, поэтому нам и кажется, что её энтропия уменьшается. Но наш демон знает о системе очень много — чтобы разделять молекулы, он должен знать положение и скорость каждой из них (по крайней мере на подлёте к нему). Если он знает о молекулах всё, то с его точки зрения энтропия системы, фактически, равна нулю — у него просто нет недостающей информации о ней. В этом случае энтропия системы как была равна нулю, так и осталась равной нулю, и второй закон термодинамики нигде не нарушился.
Но даже если демон не знает всей информации о микросостоянии системы, ему, как минимум, надо знать цвет подлетающей к нему молекулы, чтобы понять, пропускать её или нет. И если общее число молекул равно N, то демон должен обладать N бит информации о системе — но именно столько информации мы и потеряли, когда открыли перегородку. То есть количество потерянной информации в точности равно количеству информации, которую необходимо получить о системе, чтобы вернуть её в исходное состояние — и это звучит вполне логично, и опять же не противоречит второму закону термодинамики.
