Большую часть процессов, лежащих в основе химической технологии, составляют каталитические реакции. Это связано с тем, что при введении катализатора скорость взаимодействия веществ существенно увеличивается. При этом производителям удается сократить расходы или же получить большее количество продуктов реакции за тот же период времени. Именно поэтому изучению катализа уделяется много внимания при подготовке технологов. Однако это явление играет важную роль и в природе. Так, особые вещества регулируют протекание биохимических реакций в живых организмах, влияя тем самым на обмен веществ.

Понятие катализа
Суть этого химического явления заключается в регулировании скорости превращения веществ с использованием особых реагентов, способных замедлять или ускорять этот процесс. При этом говорят о положительном или отрицательном катализе. Существует также явление автокатализа, когда на скорость реакции влияет один из промежуточных продуктов химической реакции. Каталитические процессы разнообразны, отличаются они механизмами, агрегатным состоянием соединений и направлением.
Вещества, которые замедляют химические взаимодействия, называют ингибиторами, а ускоряющие каталитические реакции – катализаторами. И те, и другие изменяют скорость реакции путем многократного промежуточного взаимодействия с одним или несколькими ее участниками. При этом они не входят в состав продуктов и восстанавливаются после окончания цикла превращения веществ. Поэтому участие катализатора не отражают в уравнении реакции стехиометрически, а лишь указывают как условие взаимодействия веществ.
Виды каталитических реакций
По агрегатному состоянию веществ, принимающих участие в химической реакции, различают:
- гомогенные реакции – реагирующие вещества, продукты и катализатор находятся в одном агрегатном состоянии (фазе), при этом молекулы веществ равномерно распределены по всему объему;
- межфазные каталитические реакции – происходят на границе раздела несмешивающихся жидкостей, а роль катализатора сводится к переносу реагентов через нее;
- гетерогенные каталитические реакции – в них катализатор имеет отличное от реагентов агрегатное состояние, а сама она осуществляется на поверхности раздела фаз;
- гетерогенно-гомогенные реакции – инициируются на поверхности раздела с катализатором, а продолжаются в реакционном объеме;
- микрогетерогенные реакции – мелкие частички твердого катализатора образуют мицеллы по всему объему жидкой фазы.
Существует также окислительно-восстановительный катализ, сопровождающийся изменением степени окисления катализатора при взаимодействии с реагентами. Такие превращения называют каталитическими реакциями окисления и восстановления. Наиболее распространено в химическом производстве окисление диоксида серы до триоксида при получении серной кислоты.

Виды катализаторов
По агрегатному состоянию катализаторы бывают жидкие (H2SO4, Н3РО4), твердые (Pt, V2О5, Al2О3) и газообразные (BF3).
По типу вещества катализаторы классифицируют на:
- металлы – могут быть чистыми, сплавами, цельными или нанесенными на пористую основу (Fe, Pt, Ni, Cu);
- соединения металлов типа МmЭn – наиболее распространены оксиды MgO, Al2О3, МоО3 и др.;
- кислоты и основания – используются для кислотно-основных каталитических реакций, это могут быть кислоты Льюиса, Бренстеда и др.;
- комплексы металлов – в эту группу включают также соли переходных металлов, например PdCl2, Ni(СО)4;
- ферменты (они же энзимы) – биокатализаторы, ускоряющие реакции, идущие в живых организмах.
По специфике электронного строения различают d-катализаторы, имеющие d-электроны и d-орбитали, а также s,р-катализаторы, центром которых является элемент с валентными s и р-электронами.

Свойства катализаторов
Для эффективного использования к ним применяется довольно обширный список требований, изменяющийся для конкретного процесса. Но наиболее значимы следующие два свойства катализаторов:
- Специфичность, заключается в способности катализаторов влиять только на одну реакцию или ряд однотипных превращений и не воздействовать на скорость других. Так, платина чаще всего используется в органических реакциях гидрирования.
- Селективность, характеризуется способностью ускорять одну из нескольких возможных параллельно протекающих реакций, тем самым увеличивать выход наиболее важного продукта.
Скорость каталитической реакции
Причиной ускорения взаимодействия веществ является образование активного комплекса с катализатором, приводящее к снижению энергии активации.
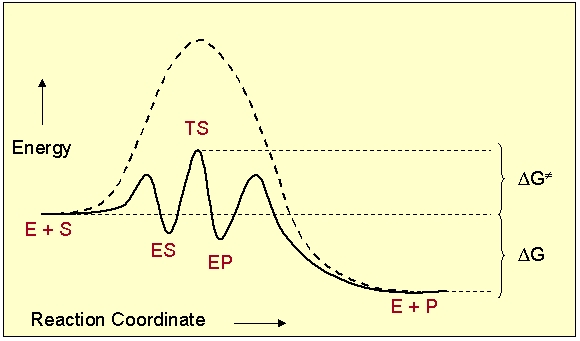
Согласно основному постулату химической кинетики, скорость любой химической реакции прямо пропорциональна произведению концентраций исходных веществ, которые взяты в степенях, соответствующих их стехиометрических коэффициентам:
v = k ∙ САх ∙ СВу ∙ СDz,
где k – константа скорости химической реакции, численно равная скорости этой же реакции, при условии, что концентрации исходных соединений равны 1 моль/л.
По уравнению Аррениуса k зависит от энергии активации:
k = A ∙ exp^(-ЕА / RT).
Указанные закономерности справедливы и для каталитических реакций. Это подтверждает и уравнение, для отношения констант скоростей:
kK / k = AК/А ∙ exp^((ЕА-ЕАК)/RT),
где переменные с индексом К относятся к каталитическим реакциям.
Стадии каталитических реакций
Для гомогенных каталитических реакций достаточно двух основных стадий:
- Образование активированного комплекса: А + К ―> АК.
- Взаимодействие активированного комплекса с другими исходными веществами: АК + В ―> С + К.
В общем виде записывают уравнение вида А + В ―> С.
Механизм гетерогенных каталитических реакций сложен. Выделяют следующие шесть стадий:
- Подведение к поверхности катализатора исходных соединений.
- Адсорбция исходных реагентов поверхностью катализатора и образование промежуточного комплекса: А + В + К ―> АВК.
- Активация образовавшегося комплекса: ΑВК ―> ΑВК*.
- Распад комплексного соединения, при этом образовавшиеся продукты адсорбированы катализатором: ΑВК* ―> CDK.
- Десорбция полученных продуктов поверхностью катализатора: CDK ―> С + D + К.
- Отвод продуктов от катализатора.
Примеры каталитических реакций
Катализаторы используют не только в химической промышленности. Любой человек в своей повседневной жизни сталкивается с различными каталитическими реакциями. Это, например, использование перекиси водорода при обработке ран. Пероксид водорода при взаимодействии с кровью начинает разлагаться под влиянием фермента каталазы:
2Н2О2 ―> О2 + 2Н2О.

В современных автомобилях система выхлопа снабжена особыми каталитическими камерами, способствующими разложению вредных газообразных веществ. Например, платина или родий помогают снижать уровень загрязнения окружающей среды оксидами азота, которые разрушаются с образованием безвредных О2 и N2.
В некоторых зубных пастах содержатся ферменты, инициирующие разложение зубного налета и остатков пищи.
Общая схема расчета кинетики гомогенных каталитических реакций
Для
представленной выше схемы каталитической
реакции легко вывести уравнение,
определяющее скорость ее протекания.
Согласно теории активированного комплеса
скорость реакции определяется скоростью
его распада на конечные продукты
![]()
.
(1)
Концентрацию
активированного комплекса (АВ)К
можно найти следующим образом. Спустя
некоторое время после начала процесса,
согласно принципу стационарности,
скорость образования активированного
комплекса будет составлять
![]()
,
(2)
откуда
![]()
.
(3)
Так
как реакция образования промежуточного
продукта АК обратима, согласно принципу
стационарности имеем
![]()
,
(4)
откуда
находим
![]()
.
(5)
Подставляя
(5) в (3) и полученное выражение в (1), находим
![]()
.
(6)
Из
уравнения (6) видно, что скорость реакции
оказывается прямо пропорциональной
концентрации катализатора, что хорошо
согласуется с опытом.
Если
константа скорости распада промежуточного
вещества на исходные продукты значительно
превышает константу скорости образования
активированного комплекса, то есть k2
>> k3
, то
![]()
.
(7)
Скорость
реакции оказывается прямо пропорциональной
концентрации катализатора и реагирующих
веществ. Промежуточное вещество при
этом называется промежуточным
веществом Аррениуса.
Если
же, наоборот, константа скорости
образования активированного комплекса
много больше константы скорости распада
промежуточного вещества, то есть k2
<< k3
, то все
количество промежуточного вещества
превращается в конечные продукты, а
скорость реакции, как видно из выражения
(6), оказывается прямо пропорциональной
концентрации того исходного вещества,
которое взаимодействует с катализатором,
образуя промежуточное вещество:
![]()
.
В
этом случае промежуточное вещество
носит название промежуточного
вещества Вант-Гоффа.
Катализ кислотами и основаниями
Многие
реакции в растворах ускоряются в
присутствии ионов водорода и гидроксила,
например, этерификация кислот и спиртов,
гидролиз сложных эфиров, инверсия
сахаров и др.
Оствальд
в 1884 г. нашел правило, согласно которому
сила каталитического действия прямо
пропорциональна электропроводности
кислот. Эта закономерность была
подтверждена Аррениусом, который, кроме
того, установил две новые (солевые
эффекты).
Прибавление
нейтральной соли, не имеющей общего
иона с катализирующей реакцию кислотой,
способно увеличивать каталитическое
действие. Например, скорость инверсии
тростникового сахара в присутствии
уксусной кислоты возрастает на 30% при
прибавлении 10% (мол.) NaCl
(первичный
солевой эффект).
Прибавление
к катализирующей реакцию кислоте ее
соли (согласно классической теории
кислот и оснований это должно уменьшать
концентрацию ионов водорода) приводит
не к уменьшению каталитического эффекта,
а в некоторых случаях даже к увеличению
(например, при этерификации трихлоруксусной
кислоты).
Прежде
чем рассматривать механизмы и кинетику
гомогенных химических реакций, ускоряемых
кислотами и основаниями, напомним
определения кислот и оснований.
Согласно
определениям, предложенным Бренстедом
и Лоури, кислота – это вещество, являющееся
донором протона, а основание – вещество,
являющееся акцептором протонов.
Кислота и основание, связанные уравнением
![]()
,
называются
сопряженными.
Протон в растворах обычно соединяется
с молекулами рстворителя. Если молекулы
растворителя не способны ни присоединять,
ни отдавать протоны, то растворенное
вещество не может проявлять ни кислотных,
ни основных свойств. Такой растворитель
называется апротонным.
Вещество только тогда может проявлять
кислотные свойства, когда растворитель
является акцептором протонов. Такой
растворитель называется протофильным.
Раствор
кислоты в воде представляет собой
систему двух пар сопряженных кислот и
оснований
![]()
.
Одна
пара сопряженных кислот и оснований –
НА и А–,
другая – Н3О+
и Н2О.
В
водных растворах оснований также имеются
две пары сопряженных кислот и оснований
![]()
![]()
.
Одна
пара сопряженных кислот и оснований
NH4+
и
NH3
, другая
Н2О
и ОН–.
Как
видно из этих примеров, вода может
выступать и как основание, и как кислота.
Вещества, способные отдавать и присоединять
протон, называются амфипротными.
Следовательно, кислотами и основаниями
могут быть не только молекулы, но и ионы.
Таким образом, в теории Бренстеда
– Лоури
основным признаком кислоты считается
наличие в ее молекуле протона. Эта теория
исключает возможность проявления
кислотного характера веществами, не
содержащими водорода, например SnCl4,
BF3
,
AlCl3
,
ZnCl2
и другими,
кислотные свойства которых хорошо
известны.
По
Льюису, кислота – вещество, способное
использовать свободную пару электронов
другой молекулы
для образования устойчивой электронной
оболочки, а основание
– вещество, обладающее свободной парой
электронов,
которая может быть использована для
образования устойчивой электронной
конфигурации другого атома. Льюисовские
кислоты и основания могут и не содержать
протонов и, следовательно, являться
апротонными. Всякое равновесие, связанное
с использованием молекулой свободной
электронной пары другой молекулы,
следует рассматривать как кислотно-основное.
В
зависимости от природы катализаторов
различают несколько типов катализа
кислотами и основаниями.
Каталитические
реакции, ускоряемые кислотами, можно
разделить на три типа:
1) специфический
кислотный катализ,
при котором активация субстрата
осуществляется ионами Н3О+
; 2) общий
кислотный катализ
с активацией субстрата любым донором
протонов, кроме Н3О+,
то есть под действием так называемых
обобщенных кислот Бренстеда; 3)
электрофильный
катализ,
когда катализаторами являются кислоты
Льюиса.
Каталитические
реакции, ускоряемые основаниями, также
можно разделить на три типа:
1) специфический
основной катализ
с активацией субстрата ионами гидроксила
ОН– ;
2) общий
основной катализ
с активацией субстрата с помощью любого
акцептора протона, кроме ОН–
, то есть
катализ, происходящий под действием
обобщенных оснований Бренстеда; 3)
нуклеофильный
катализ
под действием оснований Льюиса.
Если
в каталитической реакции одновременно
участвуют бренстедовские кислота и
основание, то такую каталитическую
реакцию называют реакцией общего
кислотно-основного катализа;
если в реакции одновременно участвуют
кислота и основание Льюиса, такую реакцию
называют реакцией электрофильно-нуклеофильного
катализа.
Когда общий кислотно-основной или
электрофильно-нуклеофильный катализ
осуществляется одновременно (синхронно)
путем тримолекулярного столкновения,
такой механизм называется пуш-пульным.
Специфический
катализ
Примером
специфического кислотного катализа
может явиться гидролиз сложных эфиров.
В сильнокислых растворах эта реакция
ускоряется только ионами гидроксония;
выражение для ее скорости имеет вид
![]()
,
где
[S]
– концентрация
субстрата, в данном случае сложного
эфира.
При
специфическом щелочном гидролизе
скорость реакции прямо пропорциональна
концентрации гидроксильных ионов, то
есть
![]()
.
Отличие
основного катализа от кислотного состоит
в том, что
активный промежуточный продукт образуется
не при внедрении, а при отрыве протона
от субстрата. Иногда он возникает при
внедрении гидроксила в реагирующую
группу субстрата.
Отнесение
реакций к специфическому кислотному
или щелочному катализу делается только
на основании вида кинетического
уравнения, а не на основании детального
механизма процесса.
По
механизму специфического кислотного
катализа протекает реакция инверсии
сахарозы, гидролиз ацеталей, гидратация
ненасыщенных альдегидов. По механизму
специфического основного катализа
протекает альдольная конденсация,
гидратация альдегидов, гидролиз сложных
эфиров.
Общий кислотный
и общий основной катализы
Общий
кислотный катализ, так же как и
специфический, связан с внедрением
протона в реагирующую часть молекулы
субстрата и с электронной атакой молекулы
воды. Отличие от механизма специфического
кислотного катализа состоит лишь в том,
что донором протона является не Н3О+,
а любая кислота НА Бренстеда. В общем
кислотном катализе часто оказывается,
что медленной стадией является не
распад, а образование катиона SH+
(S-субстрат).
По механизму общего кислотного катализа
протекает гидратация и дегидратация
альдегидов, гидролиз некоторых эфиров.
Общий
основной катализ тоже схематически
протекает так же, как и специфический.
Различие схем (помимо того, что акцептором
является не ОН–,
а обобщенное основание Бренстеда) обычно
сводится только к различию лимитирующих
стадий. Для специфического основного
катализа характерное быстрое образование
и медленный распад промежуточного
комплекса, а для общего основного
катализа характерен медленный процесс
образования промежуточного активного
аниона S–.
Примером
общего основного катализа является
разложение нитрамида в водном растворе
карбоновых кислот:
NH2NO2
NO2
+ H2O
Скорость
этой реакции в широком интервале рН не
зависит от концентрации водородных и
гидроксильных ионов. Было найдено, что
константа скорости этой реакции первого
порядка в присутствии уксусной кислоты
подчиняется уравнению
![]()
Общий
кислотно-основной катализ
Примером
общего кислотно-основного катализа
является реакция мутаротации глюкозы.
Изменение
циклической структуры, вследствие
которого достигается равновесное
значение удельного вращения, протекает
через ациклическую форму. Явление
изменения удельного вращения называется
мутаротацией.
Реакция мутаротации протекает как в
воде, так и в некоторых органических
растворителях. Она ускоряется введением
в смесь и кислот, и оснований; в зависимости
от их концентрации константа скорости
может быть определена следующим
эмпирическим уравнением:
![]()
В
некоторых органических растворителях
(пиридине, хлороформе, м-крезоле)
при полном отсутствии воды мутаротация
полностью прекращается. Это явление
объясняется следующим образом. Для
каталитической мутаротации глюкозы
необходимо одновременное присутствие
и кислоты (донора протонов), и основания
(акцептора протонов). Вода может быть и
акцептором, и донором протона, поэтому
ее каталитическое действие понятно. В
растворителях, не содержащих водорода
(апротонные растворители), может
осуществляться лишь общий кислотный
или основной катализ, так как ионы,
аналогичные ионам гидроксония и
гидроксила, при этом отсутствуют.
Однако
в смеси одной части пиридина с двумя
частями м-крезола
мутаротация протекает в 20 раз быстрее,
чем в воде. Пиридин обладает только
основными свойствами, м-крезол
– только кислотными, и в их смеси
соблюдается необходимое условие общего
кислотно-основного катализа –
одновременное присутствие кислоты и
основания.
Электрофильный
и нуклеофильный катализы
Соединения,
способные присоединяться к свободной
электронной паре – электронные акцепторы,
или кислоты Льюиса, играют роль
катализаторов в тех случаях, когда
возникающий комплекс обладает повышенной
реакционной способностью. Опыт показывает,
что апротонные кислоты катализируют
те же реакции, что и протонные кислоты,
причём активность апротонных кислот
иногда выше, чем протонных. Возникающие
комплексы имеют ионный характер и даже
диссоциированы с образованием
промежуточного активного продукта
катионного типа.
Типичными
апротонными кислотами являются
галогенидные соединения адюминия, бора,
хлорид цинка, тетрахлорид олова.
Аналогично
апротонным кислотам ведут себя иногда
ионы металлов (железа, меди, никеля).
Активный промежуточный продукт образуется
при этом в результате двухцентрового
взаимодействия иона с субстратом и
является хелатом. Субстатами могут быть
эфиры и амиды дикарбоновых кислот,
аминокислоты, некоторые кетокислоты.
Процесс во второй стадии, как и во всяком
кислотном катализе, протекает с участием
основания.
Реагируя
с водой, апротонные кислоты превращаются
в протонные, например:
Cl
AlCl3
+ H2O
Cl – Al – OH –
H+
Cl
Сила
протонной кислоты из-за сильного смещения
электрона от протона оказывается больше
апротонной. Так, из кислот Льюиса образуют
протонные бренстедовские кислоты:
BF3
+ HF
HBF4
H+
+ BF4–
AlCl3
+ HCl
HAlCl4
H+
+ AlCl4–
Соединения
со свободной элекронной парой – основания
Льюиса – в ряде реакций играют роль
основных катализаторов. Отличие
нуклеофильного катализа от общего
основного
заключается в том, что в случае основного
катализа обычно происходит отщепление
протона, а в случае нуклеофильного –
нет.
Катализ
комплексными соединениями переходных
металлов
Комплексные
соединения переходных металлов в
последнее время находят всё большее
практическое применение. Механизм их
каталитического действия представляет
интерес для понимания не только
гомогенного, но, как будет показано
ниже, и гетерогенного катализа. Большое
значение комплексы переходных металлов
играют в биологических системах.
Как
известно, к переходным металлам относятся
элементы дополнительных подгрупп
периодической системы. Эти элементы
обладают свойствами, характерными для
металлов. Они занимают средние части
длинных периодов периодической системы
от Se
до Ni,
от Y
до Pd,
от La
до
Pt. К ним же
относятся все актиноиды. В свободных
атомах этих элементов происходит
заполнение (n
– 1) d-уровней
(где n
– номер
периода). Для этих элеметов характерна
незавершённость d-оболочки
и наличие нескольких валентных состояний.
Все
переходные элементы образуют устойчивые
комплесные соединения, являясь
комплексообразователями. Лиганд
выступает в роли донора электронов, а
комплексообразователь – в роли их
акцептора. Акцепторные свойства особенно
сильно выражены у 4d-
и 5d-элементов.
Если
3d-элементы
образуют в водных растворах простые
гидратированные ионы, то 4d-
и 5d-элементы
– комплексы своих формально простых
солей. Как мы увидим ниже, эти элементы
склонны образовывать малоустойчивые
комплексные соединения за счёт
взаимодействия d-электронов
с -электронами
органических молекул. Эти неустойчивые
реакционноспособные комплексы играют
большую роль в катализе.
Интересными
катализаторами гомогенных реакций в
растворах являются карбонилы металлов.
Они представляют собой комплексные
соединения, образованные из электронодонорных
групп СО и переходных металлов. Атомы
металлов – комплексообразователей
способны присоединять такое число
электронов, какое недостаёт до построения
атома инертного газа, находящегося в
конце периода, в котором стоит данный
металл. Примером технически важной
реакции, катализируемой карбонилами
метеллов, служит оксосинтез, или процесс
гидроформилирования – метод получения
альдегидов и олефинов при взаимодествии
жидкого субстрата, соли кобальта, СО и
водорода.
В
ходе реакции из соли кобальта и СО
образуются дикобальтоктакарбонил
[Co(CO)4]2
и карбонилгидрид кобальта НСо(СО)4.
В водном растворе НСо(СО)4
полностью ионизирован, и в насыщенном
растворе (0,056 моль/л) является сильной
кислотой.
Если
сравнивать кислотные свойства
гидрокарбонилов металлов, то оказывается,
что гидрокарбонил железа близок по силе
к уксусной кислоте, гидрокарбонил никеля
не обладает кислыми свойствами, а
гидрокарбонил кобальта, как уже сказано,
является сильной кислотой. В реакции
оксосинтеза гидрокарбонил кобальта в
106
раз активнее гидрокарбонила железа, а
гидрокарбонил никеля совсем неактивен.
Олефины, благодаря наличию -электронов,
являются электронодонорными веществами,
или основаниями Льюиса.
Реакция
гидроформилирования протекает как
реакция первого порядка по олефину и
почти первого порядка по кобальту.
Скорость реакции растет с повышением
давления водорода и снижается с ростом
давления СО независимо от природы
растворителя. Кроме того, она для
-олефинов
выше, чем для олефинов с внутренней
двойной связью, так как в последнем
случае возникают стерические затруднения.
Необходимо
отметить, что механизм реакции
гидроформилирования очень сложен и не
все детали его еще установлены.
Другой
интересной и практически важной реакцией,
в которой также существенную роль играют
-комплексы,
является реакция окисления этилена до
ацетальдегида в водном растворе палладия,
так называемый вакер-процесс:
PdCl42–
+
C2H4
+
H2O
CH3CHO
+
Pd
+
4Cl–
+
2H+
Металлический
палладий легко окисляется до PdII
двухвалентной медью, а возникающая
одновалентная медь легко окисляется
воздухом до двухвалентного состояния:
4Cl–
+
Pd
+
2Cu2+
PdCl42–
+
2Cu+
4Cu+
+
O2
+
4H+
4Cu2+
+
2H2O
Таким
образом, в ходе реакции расходуются
только этилен и кислород.
Предполагают,
что процесс протекает через образование
комплекса с олефинами:
PdCl42–
+
C2H4
[C2H4PdCl3]–
+
Cl–
(а)
[C2H4PdCl3]–
+
H2O
(C2H4PdCl2
H2O)
+
Cl–
(б)
C2H4PdCl2
H2O)
+
H2O
(C2H4PdCl2OH)–
+
H3O+
(в)
Затем
происходит внутренняя перегруппировка
этого комплекса (медленная стадия):
(C2H4PdCl2OH)–
ClPdCH2CH2OH
+ Cl–
ClPdCH2CH2OH
HCl + Pd + CH3CHO
(г)
Для
такой схемы легко получить уравнение
скорости реакции, находящейся в хорошем
согласии с опытом:
![]()
,
где
К1
, К2
, К3
– константы равновесий реакции (а), (б)
и (в) соответственно.
Как
видно из схемы реакции, связи С=О возникают
путем перегруппировки в комплексе,
содержащем ОН и С2Н4
в качестве лигандов [реакция
(г)].
Хлорид
палладия кактализирует окисление
пропилена до ацетона, окисление цис–
и
транс-бутенов
до метилэтилкетона, реакцию винилирования
при взаимодействии этилена и уксусной
кислоты в присутствии PdCl2
и
Na2HPO4.
Комплексные соединения некоторых
переходных металлов катализируют также
реакции связывания азота.
Характерной
реакцией ацетиленов является гидратация,
которая катализируется солями HgII
в кислых растворах (реакция Кучерова):
![]()
Подобно
олефинам ацетилены образуют -комплексы
со многими ионами переходных металлов,
такими, как AgI,
HgII.
Возникающий
-комплекс
является активным промежуточным
продуктом реакции. Медленной стадией
реакции является внедрение воды в
комплексный ацетилен.
Важным
примером гомогенных каталитических
реакций, в которых катализаторами
являются комплексные соединения
переходных металлов и в ходе которых
лбразуются -комплексы
с олефинами, являются реакции полимеризации
олефинов. Катализаторы для полимеризации
были предложены Циглером и Натта. Их
получают из металлорганических соединений
элементов II
и III
групп (чаще
всего используют триалкилалюминий),
гидридов щелочных металлов или смешанных
алкилгидридов металлов и соединений
переходного металла (например, галогенидов
титана).
Как
видно из изложенного, в гомогенных
системах возможны каталитические
реакции гидрирования, изомеризации и
полимеризации олефинов. Такие реакции
характерны, как будет показано ниже, и
для гетерогенных каталитических реакций.
Поэтому можно предполагать, что механизм
гетерогенных каталитических реакций
в общем аналогичен механизму гомогенных
каталитических реакций. Во многих
случаях активные участки гетерогенных
катализаторов в присутствии реагирующих
веществ ведут себя аналогично комплексам,
возникающим в гомогенных системах.
Гомогенные
системы представляют большой интерес
для раскрытия детального механизма
каталитических реакций. Удобства
гетерогенных каталитических реакций,
обусловленные легкостью отделения
катализатора от реагирующих и получающихся
веществ, заменяются в гомогенном катализе
высокой селективностью протекающих
процессов. Поэтому перспективными
являются катализаторы гомогенных
систем, закрепленные на твердых телах.
Лекция 42
Гетерогенный
катализ.
Теория
промежуточных соединений
Основные
характерные черты гетерогенных
каталитических
процессов
Для
объяснения действия гетерогенного
катализатора Д.И.Менделеев предложил
теорию промежуточных соединений
(1886). Он высказал ряд интересных мыслей
о природе гетерогенного катализа. Он
указал, что свойства молекул на поверхности
раздела фаз в энергетическом отношении
отличаются от свойств молекул в объеме;
процесс удерживания молекул на поверхности
связан с выделением тепла, которое может
идти на активирование молекул; молекулы
на поверхности переходят в более
реакционноспособное состояние, поэтому
для химических процессов большое
значение имеет контакт молекул на
границе раздела фаз; реакции на границе
раздела фаз идут с большими скоростями
при невысоких температурах.
Идея
образования промежуточных соединений
развивалась далее Сабатье и рядом других
исследователей, особенно школой
Н.Д.Зелинского. Согласно этой идее,
катализатор образует с одним из реагентов
промежуточное соединение, активируя
данный реагент и облегчая реакцию.
Типичными промежуточными соединениями
являются сорбционные соединения
Pt
– H, Ni – H, Pd – H и
др.
Отсюда
следует и один
из основных принципов гетерогенного
катализа: катализатор обладает физическим
или химическим сродством к одному или
нескольким реагентам.
Например, гидрогенизационные и
дегидрогенизационные катализаторы Pt,
Pd, Ni, Cu и
другие легко сорбируют водород, образуя
с ним промежуточные соединения (палладий
даже способен растворять газообразный
водород). Гидратирующие и дегидратирующие
катализаторы, например Al2O3
или
Al2(SO4)3
,
обладают
способностью образовывать гидратного
типа соединения с водой; их действие в
реакции дегидратации спиртов, таким
образом, подобно действию серной кислоты.
Аналогичные
явления имеют место и на окислительных
катализаторах: платина и палладий легко
образуют с кислородом сорбционные
поверхностные соединения; оксид меди
(CuO)
легко
восстанавливается до металлической
меди и снова окисляется до CuO.
Ванадий образует ряд оксидов разных
степеней окисления, легко переходящих
друг в друга.
Таким
образом, важно
не только одно сродство катализатора
к реагирующему веществу, но и лабильность,
неустойчивость получающегося
промежуточного соединения,
легко вновь распадающегося при
взаимодействии со вторым реагирующим
компонентом на продукты реакции и вновь
готовый к действию катализатор. Интересным
примером подобной регенерации катализатора
является получение водяного газа на
Fe3O4
, которое
может быть изображено следующим
стехиометрическим уравнением:
![]()
Первой
стадией этого процесса является
восстановление Fe3O4
оксидом углерода:
![]()
Затем
железо, взаимодействуя с водой, дает
водород и регенерируется в Fe3O4
:
![]()
Оба
эти процесса и дают в сумме приведенное
выше стехиометрическое уравнение.
Практически
важным свойством гетерогенного
катализатора, как и всякого катализатора,
является специфичность его действия.
Различные катализаторы могут направлять
процесс по совершенно разным путям.
Так, из этанола могут быть получены семь
различных продуктов в зависимости от
выбора катализатора или условий
его применения:
|
CН2=СНСН=СН2 |
|
|
СН3СНО + Н2 |
|
|
С2Н5ОН |
С4Н9ОН |
|
СН3СООС2Н5 СН3СОСН3 + 3Н2 + СО |
|
|
(С2Н5)2О С2Н4 + Н2О |
Специфичность
действия катализатора, позволяющая
вести процесс в желательном направлении,
является одним из основных свойств,
используемых при практическом применении
катализатора в промышленности.
Избирательное каталитическое действие
по теории промежуточных продуктов
связано с образованием промежуточных
соединений разной химической природы
на разных катализаторах.
Уже
давно установлено, что присутствие в
реагирующей смеси некоторых веществ,
часто в совершенно ничтожном количестве,
способно понижать или полностью подавлять
активность катализатора. Такие вещества
получили название каталитических
ядов, а
само явление назвали отравлением
катализаторов.
Типичными каталитическими ядами являются
соединения серы (H2S,
CS2
, тиофен,
меркаптаны и др.), синильная кислота,
оксид углерода, свободные галогены (I2
,
Cl2
,
Br2
),
ртуть и
соли ртути [HgCl2
,
Hg(CN)2
],
соединения фосфора, мышьяка, свинца и
другие. Отравление катализатора
происходит вследствие сорбции яда на
поверхности катализатора, в результате
чего затрудняется доступ к ней реагирующих
веществ. Поскольку сорбция может быть
обратимой и необратимой, различают
обратимое
и
необратимое
отравление.
Например, платиновый катализатор
отравляется в присутствии CO
и
CS2
, однако
при внесении его в чистую исходную смесь
газов активность катализатора быстро
восстанавливается. При отравлениях же
H2S
и РН3
платина необратимо и полностью
дезактивируется.
Иногда
действие яда удается использовать для
направления процесса в желательном
направлении (благоприятствующее
отравление).
Так, гидрирование бензоилхлорида в
бензольном растворе над платиной
приводит через ряд последовательных
стадий к образованию толуола
![]()
![]()
Между
тем отравление платины небольшим
количеством соединений серы (например,
путем подмешивания их к исходной смеси
или употребления в качестве растворителя
неочищенного бензола) позволяет с
хорошими выходами остановить реакцию
на стадии получения ценного продукта
– бензальдегида.
Прибавление
к катализатору вещества, которое само
по себе является каталитически
недеятельным, вызывает иногда весьма
значительное повышение эффективности
процесса. Такие вещества получили
название промоторов.
Как правило, эффективность действия
промотора зависит от его количества:
наблюдаются максимумы активности при
определенном составе смешанного
катализатора. Явление промотирования
находит большое применение на практике,
позволяя повышать активность, срок
действия и избирательность катализатора.
Массивный
металлический катализатор работает
только своей поверхностью, которая
составляет, по количеству действующего
каталитически вещества, ничтожную долю
всей массы катализатора. Поэтому уже
давно стали употреблять нанесенные
катализаторы,
приготовляемые нанесением тонкого слоя
активного вещества на каталитически
недеятельный носитель. В качестве
носителей чаще всего используют вещества
с высокоразвитой поверхностью: уголь,
силикагель, алюмогель, асбест, пемзу,
кизельгур, стекло, фарфор и др. Носитель
применяют не только для экономии
катализатора (что имеет немаловажное
значение для таких катализаторов, как
Ir,
Pt, Pd, Au, Ag, Os);
он способен в небольших пределах изменять
активность катализатора, проявляя
определенный промотирующий эффект.
Кроме того, устойчивость нанесенных
катализаторов по отношению к температурному
воздействию и к отравлению ядами, как
правило, резко повышается в сравнении
с массивными металлическими катализаторами.
Нужно
отметить, что для большинства (но не для
всех) катализаторов предварительное
прогревание, необходимое для их
активирования, может привести, начиная
с некоторой, характерной для каждого
катализатора температуры, к понижению,
а при дальнейшем увеличении температуры
прогревания – и к полной потере
активности. Происходит, как говорят,
«спекание» катализатора. Носитель
препятствует спеканию, повышая срок и
температурный интервал действия
катализатора. Благодаря этому оказывается
возможным во многих случаях повышать
температуру проведения процесса, ускоряя
его и повышая выход продукта реакции.
Ката́лиз (греч. κατάλυσις от καταλύειν «разрушение») — избирательное ускорение одного из возможных термодинамически разрешённых направлений химической реакции под действием катализатора(ов), который, согласно теории промежуточных соединений, многократно вступает в промежуточное химическое взаимодействие с участниками реакции и восстанавливает свой химический состав после каждого цикла промежуточных химических взаимодействий.[1]
Термин «катализ» был введён в 1835 году шведским учёным Йёнсом Якобом Берцелиусом.
Катализа́ция (явление катализа) распространена в природе (большинство процессов, происходящих в живых организмах, являются каталитическими) и широко используется в технике (в нефтепереработке и нефтехимии, в производстве серной кислоты, аммиака, азотной кислоты и др.: большая часть всех промышленных реакций — каталитические).
Случай, когда катализатором является один из продуктов реакции или её исходных веществ, называют автокатализом.
Основные принципы катализа[править | править код]
Катализатор изменяет механизм реакции на энергетически более выгодный, то есть снижает энергию активации.
Катализатор образует с молекулой одного из реагентов промежуточное соединение, в котором ослаблены химические связи. Это облегчает его реакцию со вторым реагентом.
Катализаторы ускоряют обратимые реакции как в прямом, так и в обратном направлениях. Поэтому они не смещают химическое равновесие[2].
Типы катализа[править | править код]
По влиянию на скорость реакции катализ многие источники делят на положительный (скорость реакции растёт) и отрицательный (скорость реакции падает). В случае ингибирования цепных реакций, ингибитор расходуется в процессе реакции, поэтому данный случай нельзя считать отрицательным катализом.
Катализ бывает гомогенным и гетерогенным (контактным). В гомогенном катализе катализатор состоит в той же фазе, что и реактивы реакции, в то время как гетерогенные катализаторы отличаются фазой.
Гомогенный катализ[править | править код]
Примером гомогенного катализа является разложение пероксида водорода в присутствии ионов йода. Реакция протекает в две стадии:


При гомогенном катализе действие катализатора связано с тем, что он вступает во взаимодействие с реагирующими веществами с образованием промежуточных соединений, это приводит к снижению энергии активации.
Гетерогенный катализ[править | править код]
При гетерогенном катализе ускорение процесса обычно происходит на поверхности твёрдого тела — катализатора, поэтому активность катализатора зависит от величины и свойств его поверхности. На практике катализатор обычно наносят на твёрдый пористый носитель.
Механизм гетерогенного катализа сложнее, чем у гомогенного.
Механизм гетерогенного катализа включает пять стадий, причём все они обратимы:
- Диффузия реагирующих веществ к поверхности твёрдого вещества;
- Физическая адсорбция на активных центрах поверхности твёрдого вещества реагирующих молекул и затем их хемосорбция;
- Химическая реакция между реагирующими молекулами;
- Десорбция продуктов с поверхности катализатора;
- Диффузия продукта с поверхности катализатора в общий поток.
Примером гетерогенного катализа является окисление SO2 в SO3 на катализаторе V2O5 при производстве серной кислоты (контактный метод).
Течение реакции именно на поверхности катализатора можно продемонстрировать на опыте, в котором пластинку из платины нагревают в пламени газовой горелки, затем пламя тушат и пускают на пластинку струю газа из горелки, при этом пластинка снова раскаляется докрасна — окисление метана происходит на поверхности металла[3].
Носитель катализатора[править | править код]

Металлическая платина (показана стрелками), стабилизированная на носителе — оксиде алюминия
Носитель катализатора, иначе подложка (катализатора) (англ. carrier или support) — инертный или малоактивный материал, служащий для стабилизации на его поверхности частиц активной каталитической фазы.
Роль носителя в гетерогенном катализе состоит в предотвращении агломерации или спекания активного компонента, что позволяет поддерживать высокую площадь контакта активного вещества (см. активная каталитическая фаза) и реагентов. Количество носителя, как правило, гораздо больше количества нанесённого на него активного компонента. Основными требованиями к носителям являются большая площадь поверхности и пористость, термическая стабильность, химическая инертность, высокая механическая прочность. В ряде случаев носитель влияет на свойства активной фазы (эффект «сильного взаимодействия металл-носитель»). В качестве носителей применяют как природные (глины, пемза, диатомит, асбест и др.), так и синтетические материалы (активные угли, силикагель, алюмосиликаты, оксиды алюминия, магния, циркония и др.)[4].
Химия катализа[править | править код]
Химия катализа изучает вещества, изменяющие скорость химических реакций. Вещества, замедляющие реакции, называются ингибиторами. Ферменты — это биологические катализаторы. Катализатор не находится в стехиометрических отношениях с продуктами и регенерируется после каждого цикла превращения реагентов в продукты. Несмотря на появление новых способов активации молекул (плазмохимия, радиационное и лазерное воздействия и другие), катализ − основа химических производств (относительная доля каталитических процессов составляет 80-90 %).
Реакция, накормившая человечество (решение проблемы связанного азота) — цикл Габера-Боша. Аммиак получают с катализатором — пористым железом. Протекает при Р = 30 МПа и Т = 420—500 °C
3Н2 + N2 = 2NH3
Водород для синтеза NH3 получают путём двух последовательных каталитических процессов: конверсии СН4(СН4 + Н2О → СО + 3Н2) на Ni−катализаторах и конверсии образующегося оксида углерода (СО + Н2О → СО2 + Н2). Для достижения высоких степеней превращения последнюю реакцию осуществляют в две стадии: высокотемпературная (315—480 °C) — на Fe−Cr−оксидных катализаторах и низкотемпературная (200—350 °C) — на Cu−Zn−оксидных катализаторах. Из аммиака получают азотную кислоту и другие соединения азота — от лекарств и удобрений до взрывчатых веществ.
Различают катализы ”гомогенный, гетерогенный, межфазный, мицеллярный, ферментативный.
Энергия активации E каталитических реакций значительно меньше, чем для той же реакций в отсутствие катализатора. Например, для некаталитического разложения NH3 на N2 + Н2 E ~ 320 кДж/моль, для того же разложения в присутствии платины Е ~ 150 кДж/моль. Благодаря снижению E обеспечивается ускорение каталитических реакций по сравнению с некаталитическими.
Применение катализа в промышленности[править | править код]
Гидрирование[править | править код]
Большое число каталитических реакций связано с активацией атома водорода и какой-либо другой молекулы, приводящей к их химическому взаимодействию. Этот процесс называется гидрированием и лежит в основе многих этапов переработки нефти и получения жидкого топлива из угля (процесс Бергиуса). Производство авиационного бензина и моторного топлива из угля было развито в Германии во время Второй мировой войны, поскольку в этой стране нет нефтяных месторождений. Процесс Бергиуса заключается в непосредственном присоединении водорода к углю. Уголь нагревают под давлением в присутствии водорода и получают жидкий продукт, который затем перерабатывают в авиационный бензин и моторное топливо. В качестве катализатора используют оксид железа, а также катализаторы на основе олова и молибдена. Во время войны на 12 заводах Германии с помощью процесса Бергиуса получали примерно 1400 т жидкого топлива в сутки. Другой процесс, Фишера — Тропша, состоит из двух стадий. Вначале уголь газифицируют, то есть проводят реакцию его с водяным паром и кислородом и получают смесь водорода и оксидов углерода. Эту смесь превращают в жидкое топливо с помощью катализаторов, содержащих железо или кобальт. С окончанием войны производство синтетического топлива из угля в Германии было прекращено. В результате повышения цен на нефть, последовавшего за нефтяным эмбарго в 1973—1974, были предприняты энергичные усилия по разработке экономически выгодного способа получения бензина из угля. Так, прямое ожижение угля можно проводить более эффективно, используя двухстадийный процесс, в котором сначала уголь контактирует с алюмокобальтомолибденовым катализатором при относительно низкой, а затем при более высокой температуре. Стоимость такого синтетического бензина выше, чем получаемого из нефти.
Кислотный катализ[править | править код]
Каталитическая активность большого класса катализаторов обусловливается их кислотными свойствами. Согласно Й. Брёнстеду и Т. Лоури, кислота — это соединение, способное отдавать протон. Сильные кислоты легко отдают свои протоны основаниям. Концепция кислотности получила дальнейшее развитие в работах Г. Льюиса, который дал определение кислоты как вещества, способного принимать электронную пару от вещества-донора с образованием ковалентной связи за счёт обобществления этой электронной пары. Эти идеи вместе с представлениями о реакциях с образованием карбений-ионов помогли понять механизм разнообразных каталитических реакций, особенно тех, в которых участвуют углеводороды.
Силу кислоты можно определить с помощью набора оснований, изменяющих цвет при присоединении протона. Оказывается, некоторые промышленно важные катализаторы ведут себя как очень сильные кислоты. К ним относится катализатор процесса Фриделя — Крафтса, такой, как HCl-AlCl2O3 (или HAlCl4), и алюмосиликаты. Сила кислоты — это очень важная характеристика, поскольку от неё зависит скорость протонирования — ключевого этапа процесса кислотного катализа.
Активность таких катализаторов, как алюмосиликаты, применяющихся при крекинге нефти, определяется присутствием на их поверхности кислот Брёнстеда и Льюиса. Их структура аналогична структуре кремнезёма (диоксида кремния), в котором часть атомов Si4+ замещена атомами Al3+. Лишний отрицательный заряд, возникающий при этом, может быть нейтрализован соответствующими катионами.
Активность кислотных катализаторов обусловливается их способностью реагировать с углеводородами с образованием в качестве промежуточного продукта карбений-иона. Алкилкарбений-ионы содержат положительно заряженный углеродный атом, связанный с тремя алкильными группами и/или атомами водорода. Они играют важную роль как промежуточные продукты, образующиеся во многих реакциях с участием органических соединений. Механизм действия кислотных катализаторов можно проиллюстрировать на примере реакции изомеризации н-бутана в изобутан в присутствии HCl-AlCl3 или Pt-Cl-Al2O3. Сначала малое количество олефина С4Н8 присоединяет положительно заряженный ион водорода кислотного катализатора с образованием третичного карбений-иона. Затем отрицательно заряженный гидрид-ион Н– отщепляется от н-бутана с образованием изобутана и вторичного бутилкарбений-иона. Последний в результате перегруппировки превращается в третичный карбений-ион. Эта цепочка может продолжаться с отщеплением гидрид-иона от следующей молекулы н-бутана и т. д.
Существенно, что третичные карбений-ионы более стабильны, чем первичные или вторичные. Вследствие этого на поверхности катализатора присутствуют в основном именно они, а потому основным продуктом изомеризации бутана является изобутан.
Кислотные катализаторы широко применяются при переработке нефти — крекинге, алкилировании, полимеризации и изомеризации углеводородов. Установлен механизм действия карбений-ионов, играющих роль катализаторов в этих процессах. При этом они участвуют в целом ряде реакций, включая образование малых молекул путём расщепления больших, соединение молекул (олефина с олефином или олефина с изопарафином), структурную перегруппировку путём изомеризации, образование парафинов и ароматических углеводородов путём переноса водорода.
Одно из последних применений кислотного катализа в промышленности — получение этилированных топлив присоединением спиртов к изобутилену или изоамилену. Добавление кислородсодержащих соединений в бензин уменьшает концентрацию оксида углерода в выхлопных газах. Метил-трет-бутиловый эфир (МТБЭ) с октановым числом смешения 109 тоже позволяет получить высокооктановое топливо, необходимое для работы автомобильного двигателя с высокой степенью сжатия, не прибегая к введению в бензин тетраэтилсвинца. Организовано также производство топлив с октановыми числами 102 и 111.
Гидрирование растительного масла[править | править код]
Одна из важнейших в практическом отношении реакций гидрирования — неполное гидрирование растительных масел до маргарина, кулинарного жира и других пищевых продуктов. Растительные масла получают из соевых бобов, семян хлопчатника и других культур. В их состав входят эфиры, а именно триглицериды жирных кислот с разной степенью ненасыщенности. Олеиновая кислота СН3(СН2)7СН=СН(СН2)7СООН имеет одну двойную связь С=С, линолевая кислота — две и линоленовая — три. Присоединение водорода с разрывом этой связи предотвращает окисление масел (прогоркание). При этом повышается их температура плавления. Твёрдость большинства получаемых продуктов зависит от степени гидрирования. Гидрирование проводят в присутствии мелкодисперсного порошка никеля, нанесённого на подложку, или никелевого катализатора Ренея в атмосфере водорода высокой степени очистки.
Дегидрирование[править | править код]
Дегидрирование — это тоже важная в промышленном отношении каталитическая реакция, хотя масштабы её применения несравнимо меньше. С её помощью получают, например, стирол — важный мономер. Для этого дегидрируют этилбензол в присутствии катализатора, содержащего оксид железа; протеканию реакции способствуют также калий и какой-нибудь структурный стабилизатор. В промышленных масштабах осуществляют дегидрирование пропана, бутана и других алканов. Дегидрированием бутана в присутствии алюмохромового катализатора получают бутены и бутадиен.
Аммиак[править | править код]
Один из самых простых с химической точки зрения процессов гидрирования — синтез аммиака из водорода и азота. Азот весьма инертное вещество. Для разрыва связи N-N в его молекуле необходима энергия порядка 200 ккал/моль. Однако азот связывается с поверхностью железного катализатора в атомарном состоянии, и для этого нужно всего 20 ккал/моль. Водород связывается с железом ещё более охотно.
Основный катализ[править | править код]
Активность катализаторов обуславливается их основными свойствами. Давним и хорошо известным примером таких катализаторов является гидроксид натрия, применяющийся для гидролиза или омыления жиров при получении мыла, а один из последних примеров — катализаторы, используемые при производстве полиуретановых пластиков и пенопластов. Уретан образуется при взаимодействии спирта с изоцианатом, а ускоряется эта реакция в присутствии оснóвных аминов. В ходе реакции происходит присоединение основания к атому углерода в молекуле изоцианата, в результате чего на атоме азота появляется отрицательный заряд и его активность по отношению к спирту повышается. Особенно эффективным катализатором является триэтилендиамин. Полиуретановые пластики получают при взаимодействии диизоцианатов с полиолами (полиспиртами). Когда изоцианат реагирует с водой, ранее образовавшийся уретан разлагается с выделением CO2. При взаимодействии смеси полиспиртов и воды с диизоцианатами образующийся пенополиуретан вспенивается газообразным CO2.
Катализаторы двойного действия[править | править код]
Эти катализаторы ускоряют реакции двух типов и дают лучшие результаты, чем при пропускании реагентов последовательно через два реактора, каждый из которых содержит только один тип катализатора. Это связано с тем, что активные центры катализатора двойного действия находятся очень близко друг к другу, и промежуточный продукт, образующийся на одном из них, тут же превращается в конечный продукт на другом. Хороший результат даёт объединение катализатора, активирующего водород, с катализатором, способствующим изомеризации углеводородов. Активацию водорода осуществляют некоторые металлы, а изомеризацию углеводородов — кислоты. Эффективным катализатором двойного действия, который применяется при переработке нефти для превращения нафты в бензин, является мелкодисперсная платина, нанесённая на кислый глинозём. Конверсия таких составляющих нафты, как метилциклопентан метилциклопентан (МЦП), в бензол повышает октановое число бензина. Сначала МЦП дегидрируется на платиновой части катализатора в олефин с тем же углеродным остовом; затем олефин переходит на кислотную часть катализатора, где изомеризуется до циклогексена. Последний переходит на платиновую часть и дегидрируется до бензола и водорода. Катализаторы двойного действия существенно ускоряют риформинг нефти. Их используют для изомеризации нормальных парафинов в изопарафины. Последние, кипящие при тех же температурах, что и бензиновые фракции, ценны тем, что обладают более высоким октановым числом по сравнению с неразветвлёнными углеводородами. Кроме того, превращение н-бутана в изобутан сопровождается дегидрированием, способствуя получению МТБЭ.
Стереоспецифическая полимеризация[править | править код]
Важной вехой в истории катализа явилось открытие каталитической полимеризации-олефинов с образованием стереорегулярных полимеров. Катализаторы стереоспецифической полимеризации были открыты К.Циглером, когда он пытался объяснить необычные свойства полученных им полимеров. Другой химик, Дж. Натта, предположил, что уникальность полимеров Циглера определяется их стереорегулярностью. Эксперименты по дифракции рентгеновских лучей показали, что полимеры, полученные из пропилена в присутствии катализаторов Циглера, высококристалличны и действительно имеют стереорегулярную структуру. Для описания таких упорядоченных структур Натта ввёл термины «изотактический» и «синдиотактический». В том случае, когда упорядоченность отсутствует, используется термин «атактический».
Стереоспецифическая реакция протекает на поверхности твёрдых катализаторов, содержащих переходные металлы групп IVA-VIII (такие, как Ti, V, Cr, Zr), находящиеся в не полностью окисленном состоянии, и какое-либо соединение, содержащее углерод или водород, который связан с металлом из групп I—III. Классическим примером такого катализатора является осадок, образующийся при взаимодействии TiCl4 и Al(C2H5)3 в гептане, где титан восстановлен до трёхвалентного состояния. Эта исключительно активная система катализирует полимеризацию пропилена при обычных температуре и давлении.
Каталитическое окисление[править | править код]
Применение катализаторов для управления химизмом процессов окисления имеет большое научное и практическое значение. В некоторых случаях окисление должно быть полным, например при нейтрализации СО и углеводородных загрязнений в выхлопных газах автомобилей. Однако чаще нужно, чтобы окисление было неполным, например во многих широко применяемых в промышленности процессах превращения углеводородов в ценные промежуточные продукты, содержащие такие функциональные группы, как -СНО, -СООН, -С-СО, -СN. При этом применяются как гомогенные, так и гетерогенные катализаторы. Примером гомогенного катализатора является комплекс переходного металла, который используется для окисления пара-ксилола до терефталевой кислоты, эфиры которой служат основой производства полиэфирных волокон.
Получение этилена путём дегидродимеризации метана[править | править код]
Синтез этилена посредством дегидродимеризации позволяет превращать природный газ в более легко транспортируемые углеводороды. Реакцию 2CH4 + 2O2 → C2H4 + 2H2O проводят при 850 °С с использованием различных катализаторов; наилучшие результаты получены с катализатором Li-MgO. Предположительно реакция протекает через образование метильного радикала путём отщепления атома водорода от молекулы метана. Отщепление осуществляется не полностью восстановленным кислородом, например О2−
2. Метильные радикалы в газовой фазе рекомбинируют с образованием молекулы этана и в ходе последующего дегидрирования превращаются в этилен. Ещё один пример неполного окисления — превращение метанола в формальдегид в присутствии серебряного или железомолибденового катализатора.
Катализаторы гетерогенного окисления[править | править код]
Эти катализаторы обычно являются сложными твёрдыми оксидами. Каталитическое окисление проходит в два этапа. Сначала кислород оксида захватывается адсорбированной на поверхности оксида молекулой углеводорода. Углеводород при этом окисляется, а оксид восстанавливается. Восстановленный оксид взаимодействует с кислородом и возвращается в исходное состояние. Используя ванадиевый катализатор, неполным окислением нафталина или бутана получают фталевый ангидрид.
См. также[править | править код]
- Катализатор
- Ингибитор
- Автокатализ
- Супрамолекулярный катализ
Примечания[править | править код]
- ↑ Шмидт Ф. К. Физико-химические основы катализа — И.: Фрактал — 2004. — С. 9
- ↑ Ходаков Ю.В., Эпштейн Д.А., Глориозов П.А. § 97. Химическое равновесие // Неорганическая химия: Учебник для 7—8 классов средней школы. — 18-е изд. — М.: Просвещение, 1987. — С. 220—222. — 240 с. — 1 630 000 экз.
- ↑ Катализ // Энциклопедический словарь юного химика. 2-е изд. / Сост. В. А. Крицман, В. В. Станцо. — М.: Педагогика, 1990. — С. 103—104. — ISBN 5-7155-0292-6.
- ↑ Носитель катализатора. Дата обращения: 15 сентября 2011. Архивировано 23 декабря 2011 года.
Литература[править | править код]
- Боресков Г. К. Катализ. Вопросы теории и практики. — Новосибирск, 1987.
- Гейтс Б. Химия каталитических процессов / Б. Гейтс, Дж. Кетцир, Г. Шуйт. — М.: Мир, 1981. — 551 с.
- Журнал «Кинетика и катализ».
- Колесников И. М. Катализ и производство катализаторов. — М.: Техника, 2004. — 399 с.
- Яблонский Г. С., Быков В. И., Горбань А. Н. Кинетические модели каталитических реакций Архивная копия от 24 марта 2009 на Wayback Machine. — Новосибирск: Наука (Сиб. отделение), 1983. — 255 c.
- Клабуновский Е. И. Стереоспецифический катализ. — М.: Наука, 1968. — 367 с.
В данной статьей будут рассмотрены каталитические реакции. Читателя ознакомят с общим представлением о катализаторах и их воздействии на систему, а также будут описаны виды реакций, особенности их протекания и многое другое.
Введение в катализ

Прежде чем ознакомиться с каталитическими реакциями, стоит узнать, что же такое – катализ.
Это выборочный процесс ускорения, определенного термодинамически разрешенного направления реакции, что подвергается воздействию катализатора. Он многократно включается во взаимодействие химической природы, а влияние оказывает на участников реакции. В конце любого цикла промежуточного характера катализатор возобновляет свою изначальную форму. Введено в оборот понятие катализатора было Я. Барцелиусом и Йенсом в 1835.

Общие сведения
Катализация широко распространяется в природе и повсеместно используется человеком в технологической промышленности. Преобладающее количество всех используемых в промышленности реакций каталитические. Существует понятие об автокатализе – явлении, при котором ускоритель выступает в качестве продукта реакции либо входит в состав исходных соединений.
Все виды химического взаимодействия реагирующих веществ делятся на каталитические и некаталитические реакции. Ускорение реакций с участием катализаторов называется положительным катализом. Замедление скорости взаимодействия протекает при участии ингибиторов. Реакции носят отрицательно-каталитический характер.
Каталитическая реакция – это не только способ увеличения производительной мощи, но и возможность, повышающая качество получаемого продукта. Это обусловлено способностью специально подобранного вещества ускорить основную реакцию и замедлить скорость параллельно идущих.
Каталитические реакции также понижают затраты энергии, что расходует аппаратура. Это связано с тем, что ускорение позволяет протекать процессу в условиях более низкой температуры, которая требовалась бы без его наличия.
Примером каталитической реакции может служить получение на производстве таких ценных вещей, как: азотная кислота, водород, аммиак и т. д. Наибольшее применение эти процессы находят в производстве альдегидов, фенола, различных пластмасс, смол и каучуков и т. д.
Разнообразие реакций
Суть катализа лежит в переведении механизма протекания реакции на самый выгодный вариант. Это становится возможным благодаря снижению энергии активации.
Катализатор образовывает слабую химическую связь с определенным реагентом молекулы. Это позволяет облегчить протекание реакции с другим реагентом. Вещества, которые относятся к каталитическим, не влияют на смещение химического равновесия, так как действуют обратимо в обоих направлениях.
Катализ делится на два основных типа: гомогенный и гетерогенный. Общей чертой всех взаимодействий первого типа является нахождение катализатора в общей фазе с реактивом самой реакции. Второй тип имеет отличие в этом пункте.
Гомогенные каталитические реакции показывают нам, что ускоритель, вступая во взаимодействие с определенным веществом, образует промежуточное соединение. Это в дальнейшем приведет к снижению количества энергии, необходимого для активации.
Гетерогенный катализ ускоряет процесс. Как правило, протекает на поверхности твердых тел. Вследствие этого, возможности катализатора и его активность определяются величиной поверхности и индивидуальных свойств. Гетерогенно-каталитическая реакция имеет более сложный механизм работы, чем гомогенная. В его механизм включено 5 стадий, каждая из которых может быть обратимой.
На первой стадии начинается диффузия взаимодействующих реагентов к площади твердого вещества, далее происходит адсорбция физического характера и следом хемосорбция. Вследствие этого наступает третья стадия, при которой реакция начинает протекать между молекулами реагирующих веществ. На четвертой стадии наблюдается десорбция продукта. На пятой стадии происходит диффузия конечного вещества в общие потоки с плоскости катализатора.
Каталитические материалы
Существует понятие о носителе катализатора. Он представляет собой материал инертного или малоактивного типа, необходимый для приведения частицы, участвующей в фазе катализа, в стабильное состояние.
Гетерогенное ускорение необходимо для предотвращения процессов спекания и агломерации активных компонентов. В преобладающем ряде случаев количество носителей превышает наличие нанесенного компонента активного типа. К главному списку требований, которыми должен обладать носитель, можно отнести большую площадь и пористость поверхности, стабильность термической природы, инертность и устойчивость к механическому воздействию.
Химическая основа. Химия ускорения протекания взаимодействия между веществами позволяет нам выделить два вида веществ, а именно катализаторы и ингибиторы. Последние, в свою очередь, замедляют скорость реакции. Одной из разновидностей катализаторов являются ферменты.
Катализаторы стехиометрически не вступают в отношения с продуктом самой реакции и в конечном итоге всегда регенерируются. В современности существует множество способов влияния на процесс молекулярной активации. Однако катализ служит основой химического производства.
Природа катализаторов позволяет их разделить на гомогенные, гетерогенные, межфазовые, ферментативные и мицеллярные. Химическая реакция при участии катализатора позволит снизить затраты энергии, необходимой для ее активации. Например, некаталитическое разложение NH3 до азота и водорода потребует около 320 кДж/моль. Эта же реакция, но под воздействием платины, позволит снизить это число до 150 кДж/моль.
Процесс гидрирования

Преобладающее количество реакций с участием катализаторов базируется на активации водородного атома и определенной молекулы, что в дальнейшем приводит к взаимодействию химической природы. Данное явление называют гидрированием. Оно лежит в основе большинства этапов нефтепереработки и создания жидкого горючего из угля. Производство последнего было открыто в Германии, что обусловлено отсутствием месторождений нефти на территории страны. Создание такого топлива называется процессом Бергиуса. Заключается он в прямом соединении водорода и угля. Уголь подвергают нагреванию в условиях определенного давления и наличия водорода. Вследствие этого образуется продукт жидкого типа. Катализаторами выступают оксиды железа. Но иногда используют и вещества на основе таких металлов, как молибден и олово.
Существует и другой способ получения такого же топлива, который называют процессом Фишера-Тропша. Он состоит из двух стадий. На первом этапе уголь подвергают газификации, обрабатывая его взаимодействием паров воды и О2. Данная реакция приводит к образованию водородной смеси и оксида углерода. Далее при помощи катализаторов полученную смесь переводят в состояние жидкого топлива.
Взаимосвязь кислотности и каталитических возможностей

Каталитическая реакция – это явление, зависящее от кислотных свойств самого катализатора. В соответствии с определением по Й. Бренстеду, кислота – это вещество, умеющее отдавать протоны. Сильная кислота легко отдаст свой протон в пользование основанию. Г. Льюис определял кислоту как вещество, способное принимать на себя электронные пары от веществ-доноров и образовывать вследствие этого ковалентную связь. Две эти идеи позволили человеку определить суть механизма катализа.
Сила кислоты определяется при помощи наборов оснований, способных изменять свой цвет вследствие присоединения протона. Некоторые каталитические вещества, используемые в промышленности, могут вести себя как чрезвычайно сильные кислоты. Их сила определяет темп протонирования, а потому является очень важной характеристикой.
Кислотная активность катализатора обусловлена его способностями вступать в реакции с углеводородами, образовывая при этом промежуточный продукт – карбений иона.
Процесс дегидрирования
Каталитической реакцией является также и дегидрирование. Оно нередко используется в разных промышленных отраслях. Несмотря на то что каталитические процессы, основанные на дегидрировании, используются реже, чем реакции гидрирования, тем не менее они занимают важное место в человеческой деятельности. Примером каталитической реакции такого типа может послужить получение стирола – важного мономера. Для начала происходит дегидрирование этилбензола с участием веществ, содержащих оксид железа. Человек часто использует данное явление для дегидрирования многих алканов.
Двойное действие

Существуют катализаторы двойного действия, способные ускорять реакцию сразу двух типов. Вследствие чего приводят к лучшим результатам, в сравнении с пропусканием реагентов поочередно сквозь 2 реактора, содержащих только по одному типу катализаторов. Это обусловлено тем, что активный центр ускорителя с двойным действием пребывает в близком положении с другим таким же центром, а также с промежуточным продуктом. К хорошему результату приводит, например, объединение катализаторов, активирующих водород, с веществом, позволяющим протекать процессу изомеризации углеводорода. Активация часто осуществляется металлами, а изомеризация протекает при участии кислот.
Специфика основных каталитических реакций
Способности и эффективность катализатора обусловлены также его основными свойствами. Ярким примером может служить гидроксид натрия, который применяют в ходе гидролиза жиров для получения мыла. Такие типы катализаторов также используются в ходе производства пенопласта и пластинок из полиуретана. Уретан получают в ходе взаимодействия спирта и изоцианата. Ускорение реакции происходит при воздействии определенного основного амина. Основание присоединяется к атому углерода, содержащемуся в изоцианатовой молекуле. Вследствие этого атом азота становится отрицательно заряженным. Это приводит к повышению активности в отношении к спирту.
Полимеризация стереоспецифического характера

Важное историческое значение в истории изучения катализа имеет открытие полимеризации олефина с последующим получением стереорегулярных полимерных веществ. Открытие катализаторов, для которых характерна стереоспецифическая полимеризация, принадлежит К. Циглеру. Работы по получению полимеров, проведенные Циглером, заинтересовали Дж. Натта, который выдвинул предположение о том, что полимерная уникальность должна определяться его стереорегулярностью. Большое количество экспериментов с участием рентгеновских лучей, подвергающихся дифракции, доказали, что полимер, полученный из пропилена под воздействием катализатора Циглера, является высококристалличным. Эффект действия носит стереорегулярный характер.
Реакции подобного типа проходят на плоскости твердого катализатора, содержащего в себе металлы переходного типа, например Ti, Cr, V, Zr. Они должны находиться в неполном окислении. Уравнение каталитической реакции между взаимодействующими TiCl4 и Al(C2H5)3, в ходе которой образуется осадок, служит ярким тому примером. Здесь титан восстановлен до 3-хвалентного состояния. Такой вид активной системы дает возможность полимеризировать пропилен в обычных условиях температуры и давления.
Окисление в каталитических реакция
Каталитические реакции окисления обширно используются человеком, что обусловлено способностью определенных веществ регулировать скорость протекания самой реакции. Некоторые случаи требуют полного окисления, например нейтрализация CO и загрязнений, содержащих углеводороды. Однако подавляющее количество реакций требует неполного окисления. Это необходимо для получения в промышленности ценных, но промежуточных продуктов, что могут содержать определенную и важную промежуточную группу: COOH, CN, CHO, C-CO. При этом человек использует как гетерогенные, так и моногенные виды катализаторов.
Среди всех веществ, способных ускорять протекание химических реакций, важное место отведено оксидам. Преимущественно в твердом состоянии. Протекание окисления делится на 2 этапа. На первой стадии оксид кислорода захватывается углеводородной молекулой адсорбированного оксида. Вследствие этого происходит восстановление оксида и окисление углеводорода. Возобновленный оксид вступает во взаимодействие с О2 и возвращается к изначальному состоянию.
Темы кодификатора ЕГЭ: Классификация химических реакций в органической и неорганической химии.
Химические реакции — это такой вид взаимодействия частиц, когда из одних химических веществ получаются другие, отличающиеся от них по свойствам и строению. Вещества, которые вступают в реакцию — реагенты. Вещества, которые образуются в ходе химической реакции — продукты.
В ходе химической реакции разрушаются химические связи, и образуются новые.
В ходе химических реакций не меняются атомы, участвующие в реакции. Меняется только порядок соединения атомов в молекулах. Таким образов, число атомов одного и того же вещества в ходе химической реакции не меняется.
Химические реакции классифицируют по разным признакам. Рассмотрим основные виды классификации химических реакций.
Классификация по числу и составу реагирующих веществ
По составу и числу реагирующих веществ разделяют реакции, протекающие без изменения состава веществ, и реакции, протекающие с изменением состава веществ:
1. Реакции, протекающие без изменения состава веществ (A → B)
К таким реакциям в неорганической химии можно отнести аллотропные переходы простых веществ из одной модификации в другую:
Sромбическая → Sмоноклинная.
В органической химии к таким реакциям относятся реакции изомериза-ции, когда из одного изомера под действием катализатора и внешних факторов получается другой (как правило, структурный изомер).
Например, изомеризация бутана в 2-метилпропан (изобутан):
CH3-CH2-CH2-CH3 → CH3-CH(CH3)-CH3.
2. Реакции, протекающие с изменением состава
- Реакции соединения (A + B + … → D) — это такие реакции, в которых из двух и более веществ образуется одно новое сложное вещество. В неорганической химии к реакция соединения относятся реакции горения простых веществ, взаимодействие основных оксидов с кислотными и др. В органической химии такие реакции называются реакциями присоединения. Реакции присоединения — это такие реакции, в ходе которых к рассматриваемой органической молекуле присоединяется другая молекула. К реакциям присоединения относятся реакции гидрирования (взаимодействие с водородом), гидратации (присоединение воды), гидрогалогенирования (присоединение галогеноводорода), полимеризация (присоединение молекул друг к другу с образованием длинной цепочки) и др.

Например, гидратация :
CH2=CH2 + H2O → CH3-CH2-OH
- Реакции разложения (A → B + C + …) — это такие реакции, в ходе которых из одной сложной молекулы образуется несколько менее сложных или простых веществ. При этом могут образовываться как простые, так и сложные вещества.

Например, при разложении пероксида водорода:
2H2O2 → 2H2O + O2.
В органической химии разделяют собственно реакции разложения и реакции отщепления. Реакции отщепления (элиминирования) — это такие реакции, в ходе которых происходит отрыв атомов или атомных групп от исходной молекулы при сохранении ее углеродного скелета.
Например, реакция отщепления водорода (дегидрирование) от пропана:
C3H8 → C3H6 + H2
Как правило, в названии таких реакций есть приставка «де». Реакции разложения в органической химии происходят, как правило, с разрывом углеродной цепи.
Например, реакция крекинга бутана (расщепление на более простые молекулы при нагревании или под действием катализатора):
C4H10 → C2H4 + C2H6
- Реакции замещения — это такие реакции, в ходе которых атомы или группы атомов одного вещества замещаются на атомы или группы атомов другого вещества. В неорганической химии эти реакции происходят по схеме:
AB + C = AC + B.

Например, более активные галогены вытесняют менее активные из соединений. Взаимодействие йодида калия с хлором:
2KI + Cl2 → 2KCl + I2.
Замещаться могут как отдельные атомы, так и молекулы.
Например, при сплавлении менее летучие оксиды вытесняют более летучие из солей. Так, нелетучий оксид кремния вытесняет оксид углерода из карбоната натрия при сплавлении:
Na2CO3 + SiO2 → Na2SiO3 + CO2
В органической химии реакции замещения — это такие реакции, в ходе которых часть органической молекулы замещается на другие частицы. При этом замещенная частица, как правило, соединяется с частью молекулы-заместителя.
Например, реакция хлорирования метана:
CH4 + Cl2 → CH3Cl + HCl
По числу частиц и составу продуктов взаимодействия эта реакция больше похожа на реакцию обмена. Тем не менее, по механизму такая реакция является реакцией замещения.
- Реакции обмена — это такие реакции, в ходе которых два сложных вещества обмениваются своими составными частями:
AB + CD = AC + BD

К реакциям обмена относятся реакции ионного обмена, протекающие в растворах; реакции, иллюстрирующие кислотно-основные свойства веществ и другие.
Пример реакции обмена в неорганической химии — нейтрализация соляной кислоты щелочью:
NaOH + HCl = NaCl + H2O
Пример реакции обмена в органической химии — взаимодействие уксусной кислоты с щелочью:
CH3-CООH + KOH = CH3-CООК + H2O
Классификация химических реакций по изменению степени окисления элементов, образующих вещества
По изменению степени окисления элементов химические реакции делят на окислительно-восстановительные реакции, и реакции, идущие без изменения степеней окисления химических элементов.
- Окислительно-восстановительные реакции (ОВР) — это реакции, в ходе которых степени окисления веществ изменяются. При этом происходит обмен электронами.
В неорганической химии к таким реакциям относятся, как правило, реакции разложения, замещения, соединения, и все реакции, идущие с участием простых веществ. Для уравнивания ОВР используют метод электронного баланса (количество отданных электронов должно быть равно количеству полученных) или метод электронно-ионного баланса.
В органической химии разделяют реакции окисления и восстановления, в зависимости от того, что происходит с органической молекулой.
Реакции окисления в органической химии — это реакции, в ходе которых уменьшается число атомов водорода или увеличивается число атомов кислорода в исходной органической молекуле.
Например, окисление этанола под действием оксида меди:
CH3-CH2-OH + CuO → CH3-CH=O + H2O + Cu
Реакции восстановления в органической химии — это реакции, в ходе которых увеличивается число атомов водорода или уменьшается число атомов кислорода в органической молекуле.
Например, восстановление уксусного альдегида водородом:
CH3-CH=O + H2 → CH3-CH2-OH
- Протолитические реакции и реакции обмена — это такие реакции, в ходе которые степени окисления атомов не изменяются.
Например, нейтрализация едкого натра азотной кислотой:
NaOH + HNO3 = H2O + NaNO3
Классификация реакций по тепловому эффекту
По тепловому эффекту реакции разделяют на экзотермические и эндотермические.
Экзотермические реакции — это реакции, сопровождающиеся выделением энергии в форме теплоты (+Q). К таким реакциям относятся почти все реакции соединения.
Исключения — реакция азота с кислородом с образованием оксида азота (II) — эндотермическая:
N2 + O2 = 2NO – Q
Реакция газообразного водорода с твердым йодом также эндотермическая:
H2 + I2 = 2HI – Q
Экзотермические реакции, в ходе которых выделяется свет, называют реакциями горения.
Например, горение метана:
CH4 + O2 = CO2 + H2O

Также экзотермическими являются:
- реакции щелочных металлов с водой;
- реакции, сопровождающиеся взрывом;

- разложение дихромата аммония («вулканчик»);
- образование аммиака: N2 + 3H2 = 2NH3;
- реакции нейтрализации;
- синтез метанола;
- алюмотермия;
- реакции, в которых из менее стабильных веществ образуются более стабильные;
- в органической химии — реакции присоединения, реакции горения, окисления и др.
Эндотермические реакции — это реакции, сопровождающиеся поглощением энергии в форме теплоты (— Q). Как правило, с поглощением теплоты идет большинство реакций разложения (реакции, требующие длительного нагревания).
Например, разложение известняка:
CaCO3 → CaO + CO2 – Q
Также эндотермическими являются:
- реакции гидролиза;
- реакции, идущие только при нагревании;
- реакции, протекающие только при очень высоких температурах или под действием электрического разряда.
Например, превращение кислорода в озон:
3O2 = 2O3 — Q
В органической химии с поглощением теплоты идут реакции разложения. Например, крекинг пентана:
C5H12 → C3H6 + C2H6 – Q
Классификация химических реакций по агрегатному состоянию реагирующих веществ (по фазовому составу)
Вещества могут существовать в трех основных агрегатных состояниях — твердом, жидком и газообразном. По фазовому состоянию разделяют реакции гомогенные и гетерогенные.
- Гомогенные реакции — это такие реакции, в которых реагирующие вещества и продукты находятся в одной фазе, и столкновение реагирующих частиц происходит во всем объеме реакционной смеси. К гомогенным реакциям относят взаимодействия жидкость-жидкость и газ-газ.
Например, окисление сернистого газа:
2SO2(г) + O2(г) = 2SO3(г)
- Гетерогенные реакции — это реакции, в которых реагирующие вещества и продукты находятся в разных фазах. При этом столкновение реагирующих частиц происходит только на границе соприкосновения фаз. К таким реакциям относятся взаимодействия газ-жидкость, газ-твердая фаза, твердая-твердая, и твердая фаза — жидкость.
Например, взаимодействие углекислого газа и гидроксида кальция:
CO2(г) + Ca(OH)2(р-р) = CaCO3(тв) + H2O
Для классификации реакций по фазовому состоянию полезно уметь определять фазовые состояния веществ. Это достаточно легко сделать, используя знания о строении вещества, в частности, о типах кристаллической решетки.
Вещества с ионной, атомной или металлической кристаллической решеткой, как правило твердые при обычных условиях; вещества с молекулярной решеткой, как правило, жидкости или газы при обычных условиях.
Обратите внимание, что при нагревании или охлаждении вещества могут переходить из одного фазового состояния в другое. В таком случае необходимо ориентироваться на условия проведения конкретной реакции и физические свойства вещества.
Например, получение синтез-газа происходит при очень высоких температурах, при которых вода — пар:
CH4(г) + H2O(г) = CO(г) + 3H2(г)
Таким образом, паровая конверсия метана — гомогенная реакция.
Классификация химических реакций по участию катализатора
Катализатор — это такое вещество, которое ускоряет реакцию, но не входит в состав продуктов реакции. Катализатор участвует в реакции, но практически не расходуется в ходе реакции. Условно схему действия катализатора К при взаимодействии веществ A + B можно изобразить так:
A + K = AK;
AK + B = AB + K.
В зависимости от наличия катализатора различают каталитические и некаталитические реакции.
- Каталитические реакции — это реакции, которые идут с участием катализаторов.
Например, разложение бертолетовой соли: 2KClO3 → 2KCl + 3O2. - Некаталитические реакции — это реакции, которые идут без участия катализатора.
Например, горение этана: 2C2H6 + 5O2 = 2CO2 + 6H2O.
Все реакции, протекающие с участием в клетках живых организмов, протекают с участием особых белковых катализаторов — ферментов. Такие реакции называют ферментативными.
Более подробно механизм действия и функции катализаторов рассматриваются в отдельной статье.
Классификация реакций по способности протекать в обратном направлении
Обратимые реакции — это реакции, которые могут протекать и в прямом, и в и обратном направлении, т.е. когда при данных условиях продукты реакции могут взаимодействовать друг с другом.
К обратимым реакциям относятся:
- большинство гомогенных реакций,
- этерификация;
- реакции гидролиза;
- гидрирование-дегидрирование,
- гидратация-дегидратация;
- получение аммиака из простых веществ,
- окисление сернистого газа,
- получение галогеноводородов (кроме фтороводорода) и сероводорода;
- синтез метанола;
- получение и разложение карбонатов и гидрокарбонатов, и т.д.
Необратимые реакции — это реакции, которые протекают преимущественно в одном направлении, т.е. продукты реакции не могут взаимодействовать друг с другом при данных условиях.
Примеры необратимых реакций:
- горение;
- реакции, идущие со взрывом;
- реакции, идущие с образованием газа, осадка или воды в растворах;
- растворение щелочных металлов в воде; и др.
177
Создан на
11 января, 2022 От Admin

Классификация химических реакций
Тренажер задания 17 ЕГЭ по химии
1 / 10
Из предложенного перечня выберите два типа реакций, к которым можно отнести взаимодействие оксида натрия с водой.
1) обмена
2) гетерогенная
3) соединения
4) каталитическая
5) окислительно-восстановительная
2 / 10
Из предложенного перечня выберите два вещества, разложение которых является окислительно-восстановительной реакцией.
1) нитрат железа(III)
2) гидрокарбонат натрия
3) кремниевая кислота
4) карбонат кальция
5) перманганат калия
3 / 10
Из предложенного перечня выберите две эндотермические реакции.
1) разложение гидроксида магния
2) взаимодействие этилена с водородом
3) взаимодействие азота с кислородом
4) взаимодействие хлора с натрием
5) взаимодействие азота с водородом
4 / 10
Из предложенного перечня выберите два типа реакций, к которым можно отнести горение ацетилена в кислороде.
1) соединения
2) окислительно-восстановительная
3) эндотермическая
4) экзотермическая
5) гетерогенная
5 / 10
Из предложенного перечня выберите два типа реакций, к которым можно отнести взаимодействие гидроксида натрия с муравьиной кислотой.
1) замещения
2) диспропорционирования
3) окислительно-восстановительная
4) нейтрализации
5) обмена
6 / 10
Из предложенного перечня выберите два типа реакций, к которым можно отнести взаимодействие этанола с натрием.
1) этерификации
2) нейтрализации
3) замещения
4) соединения
5) окислительно-восстановительная
7 / 10
Из предложенного перечня выберите два вещества, реакция разложения которых является окислительно-восстановительной.
1) NН4NО2
2) Fе(ОН)3
3) NaHCO3
4) АgNО3
5) Н2SiO3
8 / 10
Из предложенного перечня выберите все типы реакций, к которым можно отнести взаимодействие этанола с натрием.
1) замещения
2) гомогенная
3) обмена
4) обратимая
5) окислительно-восстановительная
9 / 10
Из предложенного перечня выберите все типы реакций, к которым можно отнести взаимодействие водорода с парами йода.
1) замещения
2) гетерогенная
3) соединения
4) окислительно-восстановительная
5) обратимая
10 / 10
Из предложенного перечня выберите все вещества, взаимодействие которых с гидроксидом натрия является
реакцией нейтрализации.
1) оксид серы(VI)
2) сероводород
3) сульфат магния
4) серная кислота
5) сульфат аммония
Ваша оценка
The average score is 48%
