Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 31 августа 2021 года; проверки требует 71 правка.
У этого термина существуют и другие значения, см. Константа.
Константа диссоциации кислоты Ka (также известная как константа кислотности) — константа равновесия реакции диссоциации кислоты на катион водорода и анион кислотного остатка. Для многоосновных кислот, диссоциация которых проходит в несколько стадий, оперируют отдельными константами для разных стадий диссоциации, обозначая их как Ka1, Ka2 и т. д. Чем больше значение Ka, тем больше молекул диссоциирует в растворе и, следовательно кислота более сильная.
Примеры расчета[править | править код]
Одноосновная кислота[править | править код]
| Реакция | Ka |
|---|---|

|
![{displaystyle K_{{ce {a}}}={frac {[{ce {A^-}}][{ce {H+}}]}{{ce {[HA]}}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/1c21fe924187bc7d37588add5f341b1078833e09)
|
где A– — условное обозначение аниона кислоты, [HA] — равновесная концентрация в растворе частицы HA.
Двухосновная кислота[править | править код]
| Реакция | Ka |
|---|---|

|
![K_{a1} = {left[ H^+ right] left[ HA^- right] over left[ H_2A right]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2e863392d0d4cee771806c0c9d92a889419809ce)
|

|
![K_{a2} = {left[ H^+ right] left[ A^{2-} right] over left[ HA^- right]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7a8164a0d8570fb170ab8dc3ec9f1fdea61fdb28)
|
Фигурирующая в выражениях концентрация [H2A] — это равновесная концентрация недиссоциировавшей кислоты, а не изначальная концентрация кислоты до её диссоциации.
Величины pKa и pH[править | править код]
Чаще вместо самой константы диссоциации 

.
Величины pKa и pH связаны уравнением Гендерсона — Хассельбаха.
Уравнение Гендерсона — Хассельбаха[править | править код]
Преобразование уравнения[править | править код]
Пусть



|

|

|

|

|

|

|
|
Преобразуем уравнение
![{displaystyle {ce {pH}}=mathrm {p} K_{mathrm {a} }+lg left(mathrm {frac {[A^{-}]}{[HA]}} right)}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9629f69c46b4bc19ae8dc5247fdebd352c49a47e)


Можно заметить, что при 




|

|
В более кислой среде диссоциация кислоты уменьшается | ![{displaystyle [A^{-}]<[HA]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/201d1def47ab475d61324c303a92a7ebc3250a44)
|
|
|
|
Равновесие концентраций кислоты и её соли | ![{displaystyle [A^{-}]=[HA]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/031203f1a841069368f93efd675de837ca6ce1b6)
|

|

|
В более щелочной среде диссоциация кислоты увеличивается | ![{displaystyle [A^{-}]>[HA]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9ea7c1e23781e8480fab99ad497f97693b3ed434)
|
Другая связь pKa и pH[править | править код]
|
|
|
|
|
|
|
|
|
|
![{displaystyle K={[A^{-}][H^{+}] over [HA]}={[H^{+}]^{2} over [HA]}, [H^{+}]={sqrt {mathrm {K_{a}c} }}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/edbd927f4effa53ebcf3d228cb33b71ea5379589)

пример нахождения pH[править | править код]
Найти pH раствора 0,1 M Na2CO3
pKa1(H2CO3) = 6.3696
pKa2(H2CO3) = 10.3298
Решение:
Na2CO3 + H2O = NaOH + NaHCO3

откуда получаем


Значение pH > 7 означает, что соль Na2CO3 даёт щелочную среду
Константа диссоциации основания Kb[править | править код]


| Реакция | K |
|---|---|
|
|
|

|
![{displaystyle K_{b}={frac {[{mathsf {BH^{+}}}]cdot [{mathsf {OH^{-}}}]}{[{mathsf {B}}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/51e04dfbb17bc371ca2e05d7fe7a1e5be4cd9dae)
|



Константы диссоциации некоторых соединений[править | править код]
Кислотность воды pKa (H2O) = 15,74
Чем больше pKa , тем более основное соединение; чем меньше pKa , тем соединение более кислотное.
Например, по значению pKa можно понять, что спирты проявляют основные свойства (их pKa больше, чем у воды), а фенолы проявляют кислотные свойства.

Также по pKa можно установить ряд сил кислот, приведённый в российских школьных учебниках
| Название | Кислота | pKa1 | pKa2 | pKa3 |  при С = 1 моль/л, % при С = 1 моль/л, %
|
|
|---|---|---|---|---|---|---|
| Сильные
кислоты |
Йодоводородная | HI | -10 | 100 | ||
| Хлорная | HClO4 | -10 | 100 | |||
| Бромоводородная | HBr | -9 | 100 | |||
| Соляная (хлороводородная) | HCl | -7 | 100 | |||
| Серная | H2SO4 | -3 | 1.92 | 99,90 | ||
| Селеновая | H2SeO4 | -3 | 1.9 | 99,90 | ||
| Гидроксоний | H3O+ | -1.74 | 15.74 | 21 | 98,24 | |
| Азотная | HNO3 | -1.4 | 96,31 | |||
| Хлорноватая | HClO3 | -1 | 91,61 | |||
| Иодноватая | HIO3 | 0.8 | 32,67 | |||
| Средние
кислоты |
Сульфаминовая | NH2SO3H | 0.99 | 27,28 | ||
| Щавелевая | H2C2O4 | 1.42 | 4.27 | 17,69 | ||
| Йодная | H5IO6 | 1.6 | 14,64 | |||
| Фосфористая | H3PO3 | 1.8 | 6.5 | 11,82 | ||
| Сернистая | H2SO3 | 1.92 | 7.20 | 10,38 | ||
| Гидросульфат | HSO4– | 1.92 | 10,38 | |||
| Фосфорноватистая | H3PO2 | 2.0 | 9,51 | |||
| Хлористая | HClO2 | 2.0 | 9,51 | |||
| Фосфорная | H3PO4 | 2.1 | 7.12 | 12.4 | 8,52 | |
| Гексаакважелеза (III) катион | [Fe(H2O)6]3+ | 2.22 | 7,47 | |||
| Мышьяковая | H3AsO4 | 2.32 | 6.85 | 11.5 | 6,68 | |
| Селенистая | H2SeO3 | 2.6 | 7.5 | 4,89 | ||
| Теллуристая | H2TeO3 | 2.7 | 7.7 | 4,37 | ||
| Фтороводородная (плавиковая) | HF | 3 | 3,11 | |||
| Теллуроводородная | H2Te | 3 | 12.16 | 3,11 | ||
| Слабые
кислоты |
Азотистая | HNO2 | 3.35 | 2,09 | ||
| Уксусная | CH3COOH | 4.76 | 0,4160 | |||
| Гексаакваалюминия (III) катион | [Al(H2O)6]3+ | 4.85 | 0,3751 | |||
| Угольная | H2CO3 | 6.37 | 10.33 | 0,0653 | ||
| Сероводородная | H2S | 6.92 | 13 | 0,0347 | ||
| Дигидрофосфат | H2PO4– | 7.12 | 12.4 | 0,0275 | ||
| Хлорноватистая | HClO | 7.25 | 0,0237 | |||
| Ортогерманиевая | H4GeO4 | 8.6 | 12.7 | 0,0050 | ||
| Бромноватистая | HBrO | 8.7 | 0,0045 | |||
| Ортотеллуровая | H6TeO6 | 8.8 | 11 | 15 | 0,0040 | |
| Мышьяковистая | H3AsO3 | 9.2 | 0,0025 | |||
| Синильная (циановодородная) | HCN | 9.21 | 0,0025 | |||
| Ортоборная | H3BO3 | 9.24 | 0,0024 | |||
| Аммоний | NH4+ | 9.25 | 0,0024 | |||
| Ортокремниевая | H4SiO4 | 9.5 | 11.7 | 12 | 0,0018 | |
| Гидрокарбонат | HCO3– | 10.4 | 6,31*10^-4 | |||
| Иодноватистая | HIO | 11.0 | 3,16*10^-4 | |||
| Пероксид водорода | H2O2 | 11.7 | 1,41*10^-4 | |||
| Гидрофосфат | HPO42- | 12.4 | 6,31*10^-5 | |||
| Гидросульфат | HS– | 14.0 | 1,00*10^-5 | |||
| Вода | H2O | 15.7 | 21 | 1,41*10^-6 | ||
| Основания | Гидроксид | OH– | 21 | 3,16*10^-9 | ||
| Фосфин | PH3 | 27 | 0 | |||
| Аммиак | NH3 | 33 | 0 | |||
| Метан | CH4 | 34 | 0 | |||
| Водород | H2 | 38.6 | 0 |
См. также[править | править код]
- Буферные растворы
- Титрование
- Степень диссоциации
- Изоэлектрическая точка
- pH
Примечания[править | править код]
- ↑ Primchem 2002. Дата обращения: 14 октября 2021. Архивировано 23 октября 2021 года.

Светлана Радиковна Файзуллина
Эксперт по предмету «Химия»
Задать вопрос автору статьи
Константы кислотности и основности
Константа равновесия является количественной характеристикой силы кислоты или основания. Количественно кислотность вычисляют по отношению к воде. При этом мерой кислотности выступает константа равновесия реакции (константа кислотности) $Ka$. Для выражения $A ↔ B + H^+$ константа кислотности равна:
Стандартное равновесие имеет вид: $H_3O^+ ↔ H_2O + H^+$, $Ka = 1$. Тогда для любого кислотно — основного процесса:
$A_1 + B_2 ↔ A_2 + B_1$
При взаимодействии с водой любой кислоты $A$ константы равновесия соответствуют приведенным в справочной литературе значениям:
$A + H_2O ↔ H_3O^+ + B^-$
-
для многоосновных кислот, последовательно отдающих несколько протонов, первая константа кислотности всегда выше последующих;
-
для оксокислот (группы $-OH$ присоединены к одному и тому же атому) $pK_{a2}$ обычно больше $pK_{a1}$ на $5$.
Пример 1
Значения $pK_{ai}$ для ортофосфорной кислоты равны $2,12$; $7,21$; $12,67$ для $i = 1,2,3$ соответственно. То есть в водной среде преобладают разные кислотно — основные равновесия в зависимости от $pH.$
На рис. 1 отображено содержание разных ионов ортофосфорной кислоты как функция $pH$, $x_i$ – доля данного иона в смеси.
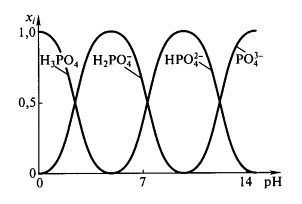
Рисунок 1. Зависимость относительного содержания различных ионов ортофосфорной кислоты от $pH$
При небольших значениях $pH$ доля иона ${PO}^{3-}_4$ очень мала, но при $pH > pKa1$ этот ион начинает доминировать. Протонированные ионы, в основном, присутствуют в растворе, когда $pH$ имеет промежуточное между соответствующими $pKa$ значение.
«Сила кислот и оснований» 👇
Константа основности рассчитывается исходя из соотношения:
$B + H_2O ↔ A +OH^-$
Константу кислотности можно связать с константой основности соотношением:
$pKa + pKb = 14.$
Чем сильнее будет сопряженная кислота, тем слабее сопряженное основание. Величине рКa соответствует значение –$lgKa.$
Кислота считается сильной, если $pKa
$HA + H_2O = +A^-$ будут идти практически до конца и кислотные свойства определяются исключительно свойствами $H_3O^+.$
Основание является в водном растворе сильным, если $pKb
Значения $pKa$ для слабых кислот лежат в интервале $sim 2-15,7$.
Автопротолиз воды
Равновесие автопротолиза воды имеет вид:
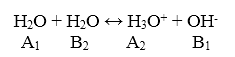
Рисунок 2.
Для данной реакции термодинамическая константа равновесия установлена экспериментально. При температуре $298 K$ она равна:
$K = [H_3O^+][OH^-] = 1 cdot 10^{-14}$
Данную константу равновесия называют ионным произведением воды $Kw.$
Согласно протолитической теории, константа равновесия реакции автопротолиза равна константе кислотности воды $Ka (H_2O).$ Исходя из величины $Kw$, определяется шкала $pH$:
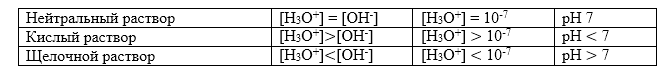
Рисунок 3.
Гидролиз
Согласно теории Бренстеда — Лоури, гидролиз солей можно рассматривать как частный случай кислотно — основного равновесия. Гидролиз — обменное взаимодействие ионов с водой, протекающее с изменением $pH$.
Гидролиз по аниону представлен уравнением:
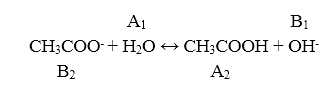
Рисунок 4.
Константа гидролиза $Kr$ является константой основности сопряженного основания $CH_3COO^-.$
Гидролиз по каниону представлен уравнением:
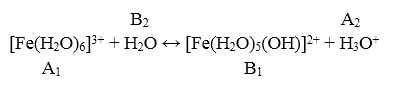
Рисунок 5.
В данном примере константа гидролиза является константой кислотности сопряженной кислоты $[Fe(H_2O)_6]^{3+}.$
Чем сильнее гидролиз по аниону, тем слабее кислота. Чем сильнее гидролиз по катиону, тем слабее основание.Равновесие гидролиза практически всегда смещено влево, при $Kr ≪ 1$. Наличие гидролиза можно определить по изменению $pH$ раствора.
Гидролиз усиливается при:
- нагревании;
- разбавлении;
- добавлении веществ, связывающих ионы $OH^-$ или $H_3O^+.$
Находи статьи и создавай свой список литературы по ГОСТу
Поиск по теме
3.2.1 Типы протолитических реакций
1. Реакции протолиза
(ионизации).
К ним относятся
реакции взаимодействия кислоты или
основания с водой:
![]()
К-та 1 осн.2 к-та 2 осн.1
![]()
К-та 1 осн.2 к-та 2 осн. 1
2. Реакции
автопротолиза,
связанные с передачей протона от одной
молекулы воды к другой.
![]()
-
Реакции гидролиза
CH3COONa+
H2O
←→
CH3COOH
+
NaOH
CH3COO–
+
H2O
←→
CH3COOH
+ OH–
осн.2 к-та 1 к-та 2
осн.1
-
Кислотно-основные
реакции
NH3
+ HCl
→
NH4+
+ Cl–
осн.2 к-та 1 к-та 2 осн.1
С точки зрения
аналитики выделяют следующие типы
реакций:
1) с переносом
протона – кислотно-основные;
2) с переносом
электрона – ОВ реакции;
3) с переносом
электронных пар с образованием связей
по донорно-акцепторному механизму –
реакции комплексообразования.
2.2.2 Константа кислотности и основности. Расчеты рН
Способность кислоты
отдавать протон, а основания принимать
его (т.е. силу кислот и оснований) можно
охарактеризовать константами равновесий,
 HS
HS
– растворитель
которые называют
константами
кислотности (Ка)
и
основности (Кb).
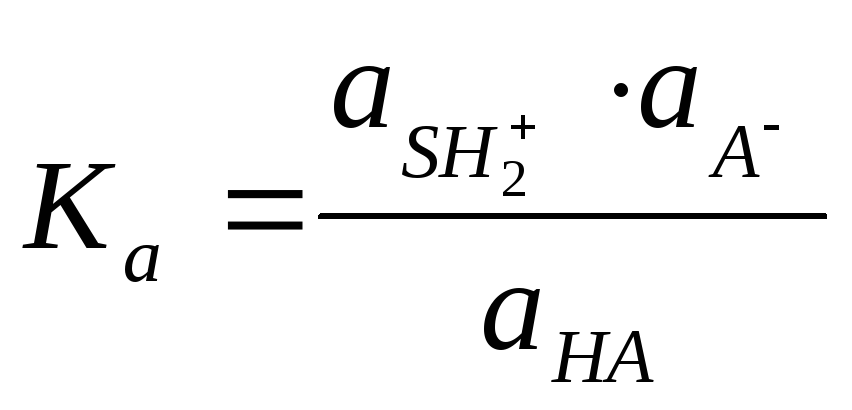
![]()
Активность
растворителя – величина постоянная
(табличные данные)
![]()
Положения
кислотно-основных равновесий
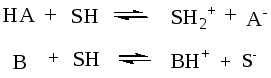
и величины
соответствующих констант кислотности
и основности зависят от природы
растворителя.
Если растворитель
– более сильный акцептор протонов, чем
вода (например, аммиак), то в нем сила
кислот возрастает. Так кислоты слабые
в водных растворах могут быть сильными
в аммиаке.
Чем сильнее
основные свойства растворителя, тем
больше кислот нивелируется в нем.
Аналогично, чем
сильнее кислотные свойства растворителя,
тем больше оснований нивелируется в
нем.
При переходе от
более к менее основному растворителю
сильные кислоты могут быть слабыми
(напр., HCl
и HClO4
в воде – сильные кислоты , а в ледяной
уксусной кислоте становятся слабыми).
Расчет рН
Расчеты
кислотно-основных равновесий используют
для:
1) нахождения рН
раствора по известным равновесным
концентрациям;
2) определения
равновесных концентраций по известному
значению рН
рН – важная оценка
для биологических жидкостей.
Для живых организмов
характерно поддержание кислотно-основного
состояния на определенном уровне. Это
находит выражение в достаточно постоянных
значениях рН биологических сред и
способности восстанавливать нормальные
значения рН при воздействии протолитов.
Система, поддерживающая
протолитический гомеостаз, включает в
себя не только физиологические механизмы
(легочную и почечную компенсации), но и
физико-химическое действие, ионный
обмен, диффузию.
В аналитической
химии важно знать концентрации всех
частиц в растворе кислоты или основания
после установления равновесия, в
частности концентрацию ионов Н+
(рН).
![]() –
–
слабый электролит
![]() –
–
сильный электролит
Чистая вода

![]()
![]()
![]()
![]()
![]()
![]()
![]()
Чистой воды не
существует. Морская вода содержит почти
все химические элементы.
Растворы слабых
кислот
![]()
![]() Т.к.
Т.к. ![]() ,
,
то
![]()
Пример 1.
Рассчитать
рН 0,1М раствора уксусной кислоты.
![]()
![]()
![]() М
М
![]() или
или
![]()
Растворы слабых
оснований
![]()
![]()
![]()
![]()
![]()
Пример 2.
Рассчитать
рН 0,1 М раствора аммония гидроксида.


![]()
![]()
![]() или
или
![]()
Растворы сильных
кислот
![]()

Для учета влияния
электростатического взаимодействия
ионов введено понятие ионной
силы раствора.
Она зависит от концентрации иона и его
заряда.
Для сильных
электролитов закон действия масс
выполняется, если пользуются активностями.
Активность учитывает концентрацию
реагентов, меж-ионное взаимодействие
(ион-ионное, ион-дипольное, диполь-дипольное,
водородные связи).
Согласно теории
Дебая и Хюккеля
![]()
![]()
![]() – зависимость
– зависимость
коэффициента подвижности от ионной
силы
А зависит от
диэлектрической постоянной растворителя
и температуры системы. При t=25°С
А=0,512 и для бинарного электролита
![]()
Пример3.
Рассчитать
рН 0,1М р-ра НСl.
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Растворы сильных
оснований
Пример 4.
Рассчитать
рН 0,1 н. раствора натрия гидроксида.
![]()
![]()
![]()
![]()
![]()
![]()
3.3
Протолитическое равновесие в буферных
растворах
В широком смысле
буферными называют системы, поддерживающие
определенное значение какого-либо
параметра при изменении состава.
Буферные растворы
могут быть кислотно-основными –
поддерживают постоянное значение рН
при введении кислот или оснований;
окислительно-восстановительными –
сохраняют постоянным потенциал системы
при введении окислителей или
восстановителей; известны металлобуферные
растворы.
Буферный раствор
представляет собой сопряженную пару;
в частности, кислотно-основной буфер –
сопряженную кислотно-основную пару:

Соседние файлы в папке Лекции
- #
- #
- #
- #
- #
- #
- #
- #
Кислотность и основность разбавленных растворов кислот и оснований
28.04.2020
Мерой силы кислоты или основания, согласно теории Брёнстеда – Лоури является константа кислотности или основности соответственно. Поскольку наиболее распространенным растворителем является вода, измерения проводят обычно в воде. Кислота в воде отдает ей свой протон:
НХ + Н2O ↔ Н3O+ + X–
Применяя закон действующих масс, получим:
В разбавленных растворах [Н2O ] = const (=55,5 моль/л), поэтому можно записать
Величина Ка называется константой кислотности (константой ионизации) и является мерой кислотности относительно стандарта, в данном случае Н2O. Константа кислотности воды равна:
Применяя аналогичный вывод для основания, получим:
Величина Кв называется константой основности и является мерой основности относительно стандарта (в данном случае Н2O ). Аналогично величине pH константы кислотности и основности можно выразить в логарифмической форме:
рКа = -lg Ка, рКв = -lg Кв (75)
Константы кислотности и основности связаны соотношением:
Kw = Ka •Kв, (76)
где Kw — константа автопротолиза растворителя (для воды — ионное произведение). Принимая во внимание (75), получаем уравнение (76) в виде:
pKw=pKa +pKв (77)
Константы кислотности Ka и pKa, pKв в воде некоторых кислот (сопряженных кислот)
|
Кислота |
Сопряженное основание |
T C |
Ka |
pKa |
pKв |
|
H3O+ |
H2O |
20 |
55,5 |
-1,7 |
15,7 |
|
HNO3 |
NO–3 |
25 |
43,6 |
-1,64 |
15,64 |
|
HNO2 |
NO–2 |
12,5 |
4,6·10-4 |
3,37 |
10,63 |
|
HBr |
Br– |
25 |
∼10 9 |
∼-9 |
∼23 |
|
HI |
I– |
25 |
∼1011 |
∼-11 |
∼25 |
|
H3PO4 |
H2PO–4 |
25 |
1,52·10-3 |
2,12 |
11,88 |
|
H2PO–4 |
HPO2-4 |
25 |
6,3·10-8 |
7,12 |
6,79 |
|
H2SO4 |
HSO–4 |
25 |
∼103 |
∼-3 |
∼17 |
|
HSO–4 |
SO2-4 |
25 |
1,2·10-2 |
1,92 |
12,08 |
|
H2S |
HS– |
18 |
9,1·10-8 |
7,04 |
6,96 |
|
H2SO3 |
HSO–3 |
18 |
1,54·10-2 |
1,82 |
12,18 |
|
H2CO3 |
HCO–3 |
25 |
4,3·10-4 |
6,37 |
7,63 |
|
HF |
F– |
25 |
3,53·10-4 |
3,45 |
10,55 |
|
HClO4 |
ClO–4 |
25 |
∼108 |
∼-8 |
∼22 |
|
HClO |
ClO– |
18 |
2,95·10-8 |
7,53 |
6,47 |
|
H2O |
HO- |
25 |
1,8·10-16 |
15,9 |
-1,9 |
|
NH+4 |
NH3 |
25 |
5,66·10-10 |
9,25 |
4,75 |
|
CH3NH+3 |
CH3NH2 |
25 |
2,7·10-11 |
10,66 |
3,34 |
|
(CH3)2NH+2 |
(CH3)2NH |
25 |
1,85·10-11 |
10,73 |
3,27 |
|
HCOOH |
HCOO- |
20 |
1,77·10-4 |
3,75 |
10,25 |
|
CH3COOH |
CH3COO– |
25 |
1,75·10-5 |
4,75 |
9,25 |
|
CF3COOH |
CF3COO– |
25 |
5,9·10-1 |
0,23 |
13,77 |
|
CH2ClCOOH |
CH2ClCOO– |
25 |
1,36·10-3 |
2,87 |
11,13 |
|
C6H5OH |
C6H5O– |
20 |
1,28·10-10 |
9,99 |
4,01 |
|
n-CH3-C6H4OH |
n-CH3-C6H4O- |
25 |
5,55·10-11 |
10,26 |
3,74 |

Выбор растворителя для измерения кислотности или основности исследуемого соединения зависит от растворимости последнего в нем. Константы автопротолиза некоторых растворителей представлены в таблице 6 -2 . Если измерения проводят в водном растворе, то
рКа = 14 – рКв (78)
Основность чаще выражается не через Кв, а через Ка сопряженны х кислот . Значения констант кислотности (Ка) и рКа, рКв, некоторых растворимых в воде кислот (сопряженных кислот) [13] приведены в таблице 6-3
Константы кислотности слабых кислот, нерастворимых в воде, определяют в других растворителях относительно друг друга, выстраивая таким образом единую шкалу кислотности. Полученные значения рКа носят приблизительный характер и могут быть использованы только для качественных оценок, например, данные таблицы 6-4 по константам кислотности относительно воды некоторых кислот Бренстеда.
Согласно табл. 6-4, в воде кислотами являются HCIO4, СН3СООН, основаниями — С2Н5ОН, С2Н2, NH3, С2Н4, С2Н6; в аммиаке кислоты — HCIO4, СН3СООН, С2Н5ОН, С2Н2, основания — С2Н4 и С2Н6.
Автор:
Источник: Органическая химия, А.М. Ким
Дата в источнике: 2002г
Кислотность и основность в водных растворах
Во всех химиях, которые вы изучали до сих пор, под кислотностью и основностью почти всегда имелось в виду определение Аррениуса – кислота это вещество, отдающее протон, а основание – гидроксид-ион. Это хорошее и правильное определение, но для органики почти бесполезное. В определении Аррениуса кислота может быть сама по себе, и основание – тоже само по себе, ведь проверяют каждое из них с помощью какой-то третьей вещи – протона или гидроксид-иона. Можно так и сказать – в этой банке кислота, а в той банке основание, а может и сама щелочь! Мало кто задумывается о том, что это возможно только в одном случае – если речь идет о водных растворах. Вода собственно и оказывает эту услугу – забирает протон у кислоты и становится ионом H3O+ гидроксония, или отдает протон основанию и становится гидроксид-ионом HO–. Гидроксоний-ион – это единый и универсальный представитель любой кислоты, ведь все кислоты только этим и различаются – сколько гидроксоний-ионов получится в растворе кислоты определенной концентрации. В воде все кислоты суть одно и то же – гидроксоний. И основания все тоже одно и то же – гидроксид-ионы. А если еще и учесть, что гидроксоний и гидроксид намертво связаны друг с другом константой диссоциации самой воды, то все становится совсем просто – есть единственный существенный показатель что кислотности, что основности – pH. А химическая природа кислоты и основания важны только для того, чтобы понять, сможем мы из них надоить нужный нам pH или нет. Мы говорим: “подкислить или подщелочить”, совершенно не концентрируясь на том, чем это сделать – любой кислотой или основанием, которое есть под рукой, лишь бы сила была достаточна. Солянкой – отлично, серной – пойдет, а у меня хлорная есть – туда ее, какая разница. А если мы в результате подкисления хотим получить слабокислый раствор, например, с pH в районе 5, то пойдет и уксусная, и фосфорная, но и те же соляная с серной, просто подкислять придется осторожно, контролируя pH индикатором или pH-метром.
Вода – это действительно уникальный растворитель, в ней все просто и элегантно, ионы в ней свободны, и все, что написано на бумаге, так и происходит в реальности, и все равны перед константой ее диссоциации. И самое главное – в водном растворе у каждой кислоты и каждого основания есть своя, одна и вполне определенная константа кислотности или основности, и они сведены в обширные и общедоступные таблицы. Все наперед известно, и любое равновесие можно взять и рассчитать.
Да, там все же есть проблема – все это хорошо только в разбавленных растворах, а в растворах с большой концентрацией наступают всякие сложности, и чем крепче раствор, тем круче сложности. В концентрированных растворах перестают соблюдаться простые законы, pH теряет смысл, и вообще начинает казаться, что кто-то подменил растворитель, и воды больше нет. На самом деле это действительно так, ведь если вы покопаетесь в памяти, то вспомните, что в корректных формулах всяких равновесий вместо концентраций присутствует какая-то загадочная термодинамическая активность, на которую все плюют, потому что в разбавленных растворах она практически не отличается от концентрации. А в концентрированных – отличается и совершенно драматически. Настолько драматически, что большинство нормальных людей предпочитает держаться подальше от концентрированных растворов. И мы так поступим. Поэтому в рассуждениях о кислотности и основности в водных растворах почти всегда избегают больших концентраций.
С проблемой корректности использования концентрация в выражениях равновесий связанао еще одно недоразумение. Довольно часто спрашивают, почему в выражениях констант кислотности или основности нет концентрации воды, и иногда даже её туда суют, думая, что исправили какой-то большой недосмотр. Но напрасно. А вот всё поэтому – потому что в корректных формулах равновесий не концентрации, а термодинамические активности, и мы договорились, что мы на это плюём, потмоу что подразумеваем, что все такие измерения проводятся в разбавленных растворах, где концентрация почти не отличается от активности, и одну можно смело заменить другой. Концентрации всех растворенных частиц – исходных электролитов и получившихся из них ионов. Но не самой воды! Вода – растворитель, ее заведомо много, поэтому ни в коем случае нельзя считать, что в выражениях констант можно заменить термодинамическую активность воды ее концентрацией. Это будет эпический провал, та самая ошибка, которая хуже, чем преступление – это насилие над самой природой констант равновесия. А в чём природа констант равновесия? В неразрывной связи с измененим стандартной энергии Гиббса, мы все знаем эту формулу, только почему-то забываем. Сами выражения констант равновесия (все эти произведения концентраций делённые на произведения других концентраций) есть не что иное как простейшее математическое следствие известного закона равновесий – в равновесии энергия Гиббса системы минимальна, а для того, чтобы найти положение равновесия, нам нужно оценить изменение стандартной энергии Гиббса правой части равновесия относительно левой части. Я специально не пишу тут никаких формул – на свете полно мест, где они выписаны, и вы сами их писали неоднократно в разных разделах физической химии. Так вот, нужно вспомнить еще одну вещь – все термодинамические функции, которые имеют размерность энергии (внутренняя энергия, энтальпия, свободная энергия, энергия Гиббса) не имеют абсолютной шкалы измерения, но отсчитываются от стандартных состояний веществ. Это такая конвенция, о которой раз и навсегда договорились, и все этим пользуются и не задают вопросов. Это произвол? Да, но общепринятый. В разных системах такими нулевыми уровнями принимают разные вещи. Например, в растворах, точкой отсчета, нулем энергии Гиббса считают чистый растворитель в стандартных условиях. А термодинамическая активность связана с энергией Гиббса, как мы знаем, логарифмической зависимостью. Вот и вся система – уровень энергии Гиббса чистого растворителя (любого!) равен нулю, а термодинамическая активность, логарифмом которой и является эта энергия, значит равна единице. Вот поэтому в выражениях констант равновесий в водных растворах и нет воды – она там есть, на самом деле, просто зачем писать параметр, величина которого равна единице. Если хотите, пишите, конечно. Именно поэтому на вопрос, какова константа диссоциации воды в воде, мы без вопросов отвечаем – это десять в минус четырнадцатой степени. И та же величина описывает и кислотность воды в воде – это и есть константа кислотности. Она же известна как ионное произведение. И pK кислотности воды в воде равна 14 – строго 14. Здесь нет вообще повода для разногласий, и любой, кто заглянет в любой хороший учебник химической термодинамики в раздел, где обсуждают константы равновесий и энергии Гиббса, немедленно в этом убедится.
В этом месте кто-нибудь обязательно возмутится и спросит – а как это, что любой растворитель задает ноль энергии Гиббса и имеет единичную активность? Но растворы ведь такие разные, это невозможно. В этом непонимание и самой химической термодинамики и вообще экспериментальной основы химии как науки. Химия изучает и описывает реальные процессы. И плевать хотела на нереальные, то есть такие, которые выходят за пределы химии. Мы ведь и для химических элементов считаем нулями стандартное состояние основного аллотропа элемента. И не паримся, что и водород, и графит, и металлическая платина – все нули. Мы не можем превратить водород в уголь, а уголь в платину. В этом месте обязательно найдется знаток космогонии, который скажет, как же не можем, а откуда тогда все взялось? Мы же знаем, что сначала был один водород (и гелий, но черт с ним, в химии гелия нет), а потом звезды сварили из водорода все остальные элементы, весьма драматическим образом. Значит можно сравнивать разные элементы по энергиям! Можно, но это не забота химии. Нам, химикам, плевать на это. Мы не звёзды, звёзды не мы. Не в этом смысле, по крайней мере, а в другом-то звёзды, конечно. В наших колбах никогда не загорится плазма, достаточная для термоядерного синтеза. Надеюсь, по крайней мере. Если кто-нибудь не уронит кузькину мать на наши лаборатории. Вот и с растворами ьакая же история, только не такая драматическая. Мы никогда не имеем дело с процессами, в которых один раствор в одном чистом расторителе в равновесном процессе преврщается в другой раствор в другом растворителе. Здесь очень важно слово “равновесном”. Попробуйте придумать такой процесс, если не верите. А пока не придумали, и не верифицировали вашу идею, придется смириться с тем, что химику совершенно до лампочки то, что в самых разных растворах в разны растворителях получаются одинаковые точки отсчета энергий Гиббса. Вообще-то это даже очень удобно. Основоположники химической термодинамики ох и неглупые были люди, прямо изумление берет какие неглупые!
Но в органической химии проблем гораздо больше. В органической химии очень редко используют воду как растворитель, особенно как единственный растворитель (довольно часто можно встретить смеси воды с другими растворителями, но почти никогда – чистую воду).
 Мы привыкли называть ион H3O+ гидроксоний-ионом. Но это устаревший термин. Он существует так давно, и мы так к нему привыкли, что кажется, что как собственно ещё может называться такой ион. Но в этом-то и дело, что так он называться не может. Окончание -оний принято лепить к названию того соединения (класса соединений), который был протонирован. Названию, конечно, не русскому, а латинизированному, и немного обчекрыженному для удобопроизносимости. Аммиак – аммоний, фосфин – фосфоний, сульфид – сульфоний, и так далее. В этой логике гидроксоний – это протонированный гидроксил, но протонированный гидроксил это просто вода, даже не ион.
Мы привыкли называть ион H3O+ гидроксоний-ионом. Но это устаревший термин. Он существует так давно, и мы так к нему привыкли, что кажется, что как собственно ещё может называться такой ион. Но в этом-то и дело, что так он называться не может. Окончание -оний принято лепить к названию того соединения (класса соединений), который был протонирован. Названию, конечно, не русскому, а латинизированному, и немного обчекрыженному для удобопроизносимости. Аммиак – аммоний, фосфин – фосфоний, сульфид – сульфоний, и так далее. В этой логике гидроксоний – это протонированный гидроксил, но протонированный гидроксил это просто вода, даже не ион.
А этот ион – это протонированная вода, вода – корень -гидро-, а не -гидрокси-. Поэтому ион этот правильно называется гидроний, и в современной литературе вы его найдёте именно под этим называнием. Название гидроний утверждено ИЮПАК, а значит является системным, а название гидроксоний – несистемным. Имейте это в виду. Можете продолжать называть этот ион гидроксонием, но не удивляйтесь, что в современных статьях такого не найдёте. Поэтому лучше переучиваться. Я потихоньку тоже буду переходить на этот термин. Раньше, еще не так давно их применяли вперемешку, и можно было не париться, но в современных журналах случилась такая жёсткая зачистка от несистемного названия, что я перепугался, и решил тоже начать переучиваться.
Почему вода – плохой растворитель в органической химии
Сразу заметим – плохой не значит бесполезный. В органике используют воду, только нужно хорошо понимать, когда это можно делать, и какие проблемы придется преодолеть.
Первая и очень важная проблема – в воде почти не растворимо подавляющее большинство органических веществ, а реакции все же принято вести в растворах. Кто-нибудь обязательно возразит – в реальной химии полно гетерогенных реакций. Да просто если обратить внимание на то, что мы делаем в практикуме, нельзя не заметить, что в каждой второй реакционной смеси что-нибудь валяется на дне или плавает, или два слоя в жидкости – ну явно это все не кристально прозрачные настоящие растворы! Но, уверяю вас, реакции идут только тогда, когда реагирующие молекулы могут встретиться, а встретиться молекулам проще всего, если они находятся в одной фазе, сохраняя при этом подвижность. Идеальный тип фазы для этого – жидкость. В газовой фазе молекулы тоже носятся как угорелые, но их там мало и у них много проблем с тем, что в газовой фазе неоткуда взять необходимую энергию, а еще чаще некуда девать лишнюю энергию, поэтому мы не будем рассматривать реакции в газовой фазе. А в твердой фазе молекулы фактически неподвижны. Поэтому реакции в подавляющем большинстве случаев (с нашей точки зрения это означает всегда) идут в жидком растворе, и газообразные или твердые вещества вначале обязательно растворяются. Очень часто растворение происходит не полностью (поэтому реагенты в химии часто берут в избытке), или происходит постепенно по мере расходования в реакции, поэтому мы и видим гетерогенные реакционные смеси. Но какая-то растворимость быть должна, и не очень малая, так как в этом случае реакция будет очень медленной. Вот в воде растворимость очень многих органических соединений настолько мала, что реакции идут, если идут, очень медленно.
Но это не все проблемы воды. Вода еще и весьма реакционноспособное соединение, и многие вещества, используемые в органике – сильные основания, гидриды, щелочные металлы, соединения, склонные к гидролизу – с водой реагируют, и мы получим не ту реакцию, которую хотели сделать. И в придачу иногда получим большие проблемы.
Вода жестко ограничивает диапазон основности и кислотности – многие органические реакции требуют более сильных кислот и оснований, чем те, которые достижимы в водных растворах.
Поэтому вместо воды приходится использовать органические жидкости, такие как эфир, ацетон, этанол, уксусную кислоту, бензол, хлороформ, и прочая, и прочая – многие десятки наименований. Это решает проблему растворимости, но создает новую проблему там, где в реакциях ожидается образование ионов. Органические жидкости малополярны и плохо сольватируют ионы. Поэтому ионы в них несвободны, они не могут отойти друг от друга: написать их на бумаге можно, но в реальности их нет, вместо них всякие ионные пары или ионные агломераты, с которыми нельзя обращаться так же, как со свободными ионами. Кроме того, если вы возьмете то, что в обычном смысле точно является кислотой, например, серную кислоту, и растворите ее в хлороформе, серной кислоте некому будет отдать протон, хлороформу он с приплатой не нужен. А если растворите в этаноле, будет еще хуже, так как пойдут всякие неожиданные реакции, явно более сложные, чем перемещение протона. И так почти везде. Что же, серная кислота уже и не кислота?!
Кислоты и основания по Бренстеду-Лоури
В неводных средах от удобной теории кислот и оснований Аррениуса придется отказаться. Вместо нее придется использовать теорию, в которой кислоты и основания выясняют между собой отношения (кислотно-основные отношения) прямо и без посредников. Может показаться, что так должно быть даже проще, но, увы, это не так, и с этой горькой истиной нам придется не только смириться, но и научиться обращаться не менее ловко.
Еще раз зададим этот странный вопрос: серная кислота – это кислота? Ответ будет не менее странным, чем вопрос: это заранее неизвестно. Или даже еще точнее – этот вопрос не имеет смысла по отношению к конкретному веществу, даже если в его названии зазывно звучит слово “кислота” (или “основание”). Кислотно-основные свойства выясняются всегда в парах веществ, тогда одно может стать кислотой, а другое основанием. А в другой паре роли вполне могут измениться на противоположные. Можно даже сказать уже совсем злобно – каждая кислота однажды станет основанием. А каждое основание кислотой. К этому парадоксу стоит отнестись с осторожностью, но первая половина его (“каждая кислота…”) почти безусловно верна, хотя вторая (“каждое основание…”) имеет огромное количество исключений, поэтому точнее можно было бы сказать: “некоторые основания могут быть и кислотами”.
Не будем авансом расстраиваться. Серная кислота почти всегда, безусловно, кислота. И это очень легко установить. Добавьте (мысленно!) в тот же раствор какое-нибудь известное основание, например, метиламин. И вы увидите, как протон от кислоты переместится к метиламину. И это произойдет в любом растворителе (даже в воде, но в воде это происходит не прямо, а через посредничество воды).
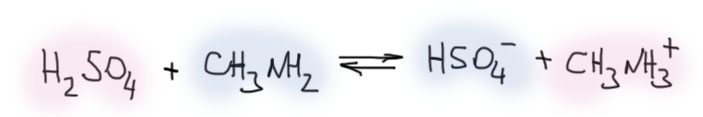
Кислота передает протон основанию. Протон не гуляет сам по себе в виде какого-то третьего иона типа гидроксония, а переходит от кислоты к основанию. Получается нечто принципиально отличающееся от Аррениуса: кислота и основание не имеют смысла сами по себе, а только в реакции друг с другом. И почти любое вещество может быть и кислотой (для этого в составе должен быть хотя бы один протон, и неважно, насколько прочно он сидит на своем месте), и основанием (для этого желательно, чтобы в молекуле был бы хотя бы один атом с неподеленной парой – желательно, но не обязательно: метан, этилен, бензол, как мы скоро увидим, вполне себе основания). А перейдет ли протон от кислоты к основанию, и насколько перейдет (то есть, какой будет константа равновесия) определяется количественными характеристиками кислоты и основания, о чем речь пойдет ниже.
В этом состоит фундаментальная концепция кислот и оснований Бренстеда-Лоури, или, как говорят и пишут гораздо чаще, просто Бренстеда (точнее, Брёнстеда), что несправедливо, но так уж устроена наука – вторых всегда забывают, и если у вас фамилия не на А или Б, или хотя бы Ц, бросайте химию и вообще науку сразу, у вас нет шансов остаться в памяти потомков.
Взаимодействие кислоты и основания – это всегда равновесие – равновесие переноса протона. Не забывайте писать знак равновесия, когда записываете реакцию между кислотой и основанием. Это – важный элемент культуры (может ли кто-нибудь признаться в желании быть диким химиком? – но если вы не пишете знак равновесия в переносе протона, то фактически признаетесь именно в этом, поздравляю), дань корректности, и элемент самодисциплины – не забывая об этом никогда, вы перестанете делать глупые ошибки, связанные с неверной оценкой относительной кислотности и основности. Почему это так важно? Да потому, что если мы ставим простую стрелку или знак равенства, то мы фактически подразумеваем, что любое взаимодействие кислоты и основания приводит к количественному переносу протона. Но это не так – перенос протона всегда равновесен, и для того, чтобы понять, насколько хорошо и насколько полно нам удалось оторвать протон от какой-то кислоты, нам нужно иметь представление об относительной кислотности и основности, и искать количественные характеристики кислот и оснований. Знак равновесия все время напоминает нам не забывать об этом.
- Кислоты Бренстеда-Лоури часто называют просто кислотами Бренстеда, или протонными кислотами, или донорами протона. Эти понятия – синонимы. Можно употреблять любое, или вперемешку разные для украшения речи.
- Основания Бренстеда-Лоури называют также основаниями Бренстеда, или акцепторами протона, но не протонными основаниями.
Никогда не путайте основания Бренстеда и основания Льюиса несмотря на то, что основания Бренстеда почти всегда являются основаниями Льюиса. Но эти два свойства проявляются в совершенно разных реакциях, а название должно соответствовать ситуации. Кислоты Бренстеда и кислоты Льюиса и так никто не путает.
Константы кислотности и основности в воде
Еще раз повторим – в уже изученных химиях (неорганической и особенно аналитической) мы имели дело преимущественно с кислотами и основаниями в водных растворах. Мы привыкли, что каждая кислота и основание характеризуется константой кислотности или основности (несколькими константами, если диссоциация по нескольким ступеням), и это именно константы, то есть постоянные величины из справочника, которые однозначно и количественно характеризуют способности кислоты и основания. Знаем константы, и больше ничего не нужно – всегда все рассчитаем, и pH среды в растворах, в том числе буферных, и всякие константы равновесий. Иными словами, пока мы не выходим за пределы разбавленных водных растворов, константы кислотности основности имеют абсолютный характер – их знание гарантирует нам правильность количественных оценок всяких равновесий в растворах. Что это значит, мы поймем только разобравшись с кислотностью и основностью по Бренстеду-Лоури.
Как только мы переходим к теории Бренстеда-Лоури (а другой в неводных растворах нет, про кислоты и основания Льюиса поговорим отдельно, но кислотность по Льюису никогда не используется для количественных оценок) эта определенность исчезает. Константы кислотности и основности по Бренстеду-Лоури всегда относительны, и шкалы кислотности и основности в неводных средах фактически являются плавающими, не привязанными к какой-то системе координат с определенным началом (в водных растворах шкалы привязаны к константе кислотности воды).
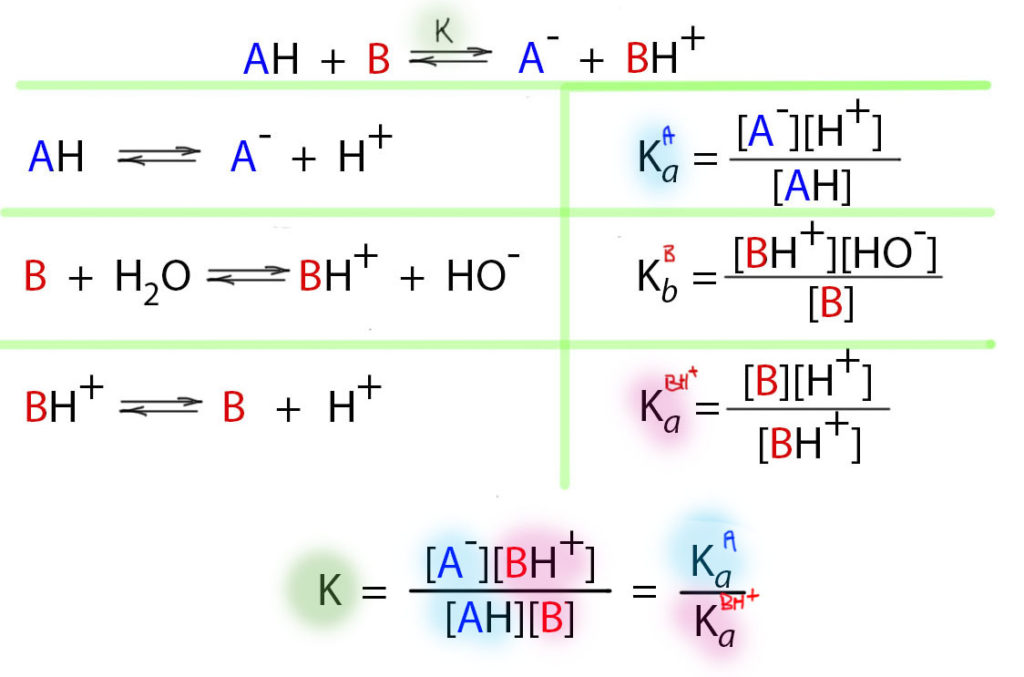
И вот почему. Напишем реакцию между кислотой и основанием сначала в воде. В воде кислота имеет константу кислотности, которая записывается очевидным образом. Основание имеет константу основности, которая также записывается вполне определенным, хотя и чуть более сложным способом, так как нужно учесть образование гидроксид иона из воды. Если перемножить константу кислотности кислоты на константу основности основания получим константу равновесия реакции кислоты и основания, но помноженную на константу кислотности воды (которую обычно называют ионным произведением, величину которого знают все). Это просто, но не удобно. Поэтому очень часто вместо константы основности основания используют константу кислотности протонированного основания (сопряженной кислоты основания).
- Тогда мы получаем очень простое правило: константа равновесия реакции кислоты и основания является частным двух констант кислотности – вверху та кислота, что стоит слева в равновесии, внизу – та, что справа (сопряженная кислота основания).
Фактически получаем просто сравнение двух кислот, и равновесие будет сдвинуто в сторону более слабой кислоты. Ровно так и должно быть “из общих соображений”, и это славно. Это легко понять и запомнить. Гораздо сложнее было бы сравнивать константу кислотности и константу основности – как понять из этих величин, куда пойдет протон? Придется в уме или на бумажке производить вычисление, домножая еще и на ионное произведение воды. Да, это нехитрое вычисление, но ум нам дан не для того, чтобы забивать его всякой дрянью.
Кислотность и основность в неводной среде
А теперь уйдем из воды. Задача та же – понять, перейдет ли протон от какой-нибудь кислоты к какому-нибудь основанию.
Сразу возникает вопрос, почему бы не использовать константы кислотности и основности из таблиц для водных растворов. Ответ простой – этого нельзя делать ни к коем случае! Растворитель очень сильно влияет на кислотность и основность. Константы кислотности из таблиц для воды совершенно не годятся. Почему бы тогда не найти таблицы кислотности и основности для других растворителей. Ответ многих должен просто шокировать. Во-первых, потому что таких таблиц для большинства (подавляющего большинства = почти всех) растворителей просто нет в природе. Чем же, черт возьми, занимаются чертовы химики, если такой простой вещи до сих пор не сделали! Чем занимаются, тем и занимаются, и если вам кажется, что не тем, займитесь этим сами. И вещь эта совсем не такая простая. Чертовски сложная это вещь на самом деле.
Попробуем прикинуть то, что мы делали для водных растворов, для произвольного растворителя. И сразу поймем, в чем одна из проблем. В водном растворе сама вода выполняет роль переносчика протона: молекула воды забирает протон у кислот и передает его основанию. Основание отнимает протон у молекулы воды, и образующийся гидроксид ион отнимает протон у кислоты. Кроме того мы знаем, что вводном растворе все ионы свободны, могут перемещаться туда, куда хотят, и единственное ограничение, которое на все это накладывается, это сохранение общей электронейтральности любого конечного объема раствора.
В других растворителях это не так. Да,есть растворители, сильно похожие на воду, например, спирты. Молекула спирта тоже может служить переносчиком протона, передавая его от кислоты к основанию почти так же, как вода. Но ионы в спиртах несвободны, катионы и анионы не могут плавать сами по себе, но обычно держатся рядом друг с другом (это называется “ионная пара”). Немного позже мы поговорим подробнее о свойствах растворителей, и попробуем разобраться, почему так происходит. Сейчас просто поймем, что это так. Почему это важно? Легко понять. Вот возьмем, например, гидроксид натрия и гидроксид калия. В воде это одно и то же основание – гидроксид-ион, а катион металла нам интересен только тогда, когда мы считаем концентрацию раствора и берем навеску реактива. Раствор 40 грамм NaOH в литре воды это с точки зрения основности точно то же самое, что раствор 56 грамм KOH в том же литре. И если представить себе ситуацию, что от вас потребовался литр одномолярной щелочи, а по недосмотру едкого натра осталось в банке на донышке всего 20 грамм, то смело растворяйте их и бросайте туда же еще 28 грамм едкого кали – никто не заметит, что это два реактива в растворе, а не один!
Но почти в любом другом растворителе, включая спирты, это – два разных основания! И если в методике написано растворить KOH в этаноле, а у вас нет едкого кали, и рука тянется за едким натром, остановите ее (руку) и идите искать калийную щелочь. Иначе с хорошей долей вероятности запорете синтез и получите плохой выход, а то и вообще не то, что ожидали. Потому что в неводных растворах катионы металлов не уходят от гидроксид-иона, взаимодействуют с ним, мешают ему реагировать с кислотой, отрывая от нее протон. Более мелкий катион натрия взаимодействует сильнее и мешает сильнее, чем более крупный катион калия (надеюсь, понятно почему – это простое следствие закона Кулона – сила притяжения зарядов очень быстро уменьшается с увеличением расстояния между центрами заряженных, в данном случае, шариков). На это все еще и наложится разная растворимость ионных веществ в разных растворителях. Не одна беда, а много разных бед! Учесть все это задача настолько сложная, что о количественных измерениях проще забыть сразу. Потом когда-нибудь разберемся. Химии ведь всего около 200 лет, пройдет еще хотя бы столько же, разберутся. А нам-то что делать? Просто не забывать, что это так, и пока поедем дальше. Еще не все проблемы выявлены.
Протон в неводных средах
В других растворителях, например, эфире, ацетоне, хлороформе протон перемещается от кислоты к основанию без помощи растворителя. Можно сказать – ну, так это проще, наверное. Кислота-основание составляют пару, которая улаживает свои дела без посредников, без воды. Но получается довольно скверная проблема. Мы больше не можем написать отдельные константы кислотности и основности. В воде это получалось только потому, что константа кислотности (или основности) в воде, это, на самом деле константа равновесия взаимодействия кислоты с водой или основания с водой. Вода везде одна и та же, поэтому мы можем отдельно измерить все эти константы, а потом использовать их для оценки равновесий. Когда в реакциях в водной среде мы пишем протон, в реальности мы имеем в виду протонированную молекулу воды (гидроксоний-ион), и это настолько хорошо все знают и понимают (надеюсь), что для простоты пишут протон.
Но, и это нужно запомнить как важнейшую и строжайшую заповедь
– не пиши протона в неводных средах твоих –
(извиняюсь за хамский тон, но в заповедях положено тыкать, типа, не убий, не прелюбодействуй, не возжелай вола, не вари козленка и т.п.).
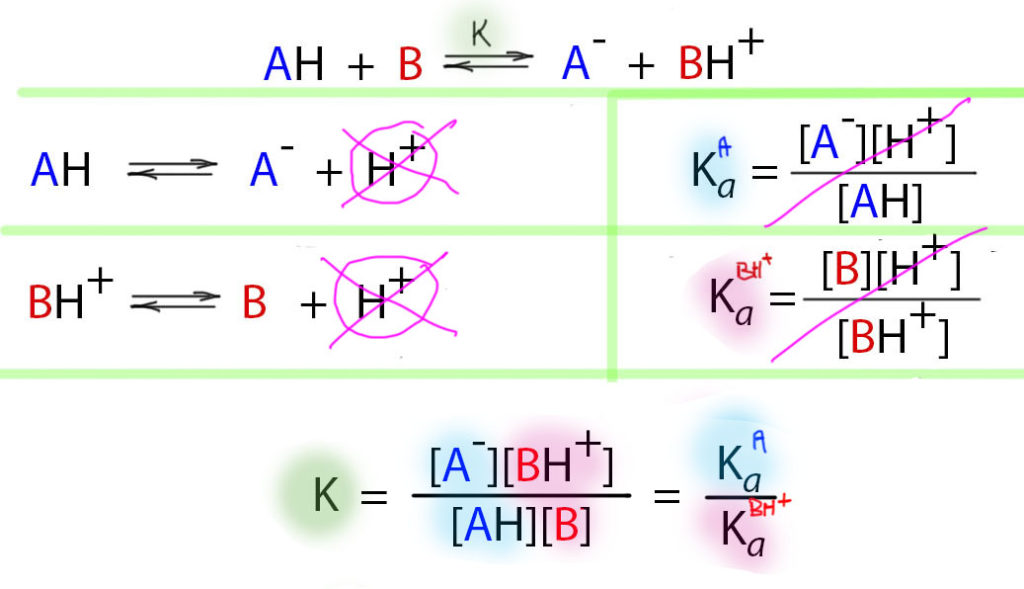
- Почему нельзя писать протон? Потому что протон – это очень маленькая заряженная штуковина (частица, обладающая явно выраженными квантовыми свойствами, и только этого нам и не хватало!), которая очень сильно взаимодействует с любыми молекулами, и в присутствии других молекул обязательно вступает с ними во взаимодействие, протонирует их. Свободный протон – невероятно, просто фантастически сильная кислота, заведомо более сильная, чем любая молекула, с которой мы можем встретиться в органической химии, в частности, чем любая молекула любого растворителя. Протон – заведомо самая сильная протонная кислота. В растворах свободный протон образоваться не может просто потому что в любом растворе обязательно найдется что-нибудь, что можно запротонировать, и писать его поэтому не стоит.
- хорошо, а почему в других растворителях нельзя, а в воде можно? Потому что мы знаем, что и в воде нет протона, а есть протонированная вода, гидроксоний-катион (и другие подобные частицы), и мы гарантируем всем, что мы это знаем, и что именно это и имеем в виду, когда в воде пишем как бы свободный протон, а на самом деле подразумеваем на уровне безусловного инстинкта, что это гидроксоний. Если вам не кажется, что это так очевидно, и вы не доверяете своим инстинктам, то не пишите протон и в воде, а пишите всегда гидроксоний-ион.
- отлично, а почему бы тогда нам не писать в других растворителях вместо протона протонированный ион растворителя? А потому что в подавляющем большинстве случаев это неверно. Во-первых, большинство растворителей слабые и очень слабые основания, которые просто не принимают протон от большинства кислот кроме самых-самых сильных. Во-вторых, для очень многих органических растворителей протонирование приводит к немедленным химическим реакциям, часто довольно сложным и малопредсказуемым. И вместо простой реакции кислота-основание мы получили бы кашу (органики называют это осмолением, то есть реакциeй, которую проще выбросить, чем разбираться, что в ней получилось).
Константы кислотности и основности в неводных средах
Итак, возвращаемся к константам кислотности основности в неводной среде. Точно так же, как в воде, пытаемся оценить равновесие реакции кислоты и основания. Вроде бы все похоже, но поскольку протон переносится непосредственно от кислоты к основанию, и мы не можем написать ни свободный протон, ни какой-то переносчик, то мы не можем написать полноценное равновесие, в котором участвовала бы только кислота или только основание, протсо потому что как только мы убираем протон, мы не можем по отдельности уравнять ни равновесие кислота – сопряженное основание, ни основание – сопряженная кслота (и там, и там протона не хватает). А значит нет больше выражений констант кислотности и основности через концентрации. И это был бы просто чертовски обидный тупик, потому что это означало бы, что нам пришлось бы по отдельности измерять и сводить в таблицы константы равновесия для каждой пары кислота-основание. Что это означает понять довольно просто на примере: вот, предположим у нас есть 10 кислот и 10 оснований. Для того, чтобы предсказать их реакции друг с другом в воде, понадобится 10 констант кислотности и 10 констант основности (или кислотности сопряженных кислот) – 20 чисел. А не в воде нам пришлось бы измерить все сто констант равновесий – во-первых, замучились бы измерять, во-вторых, это уже не наука, а спорт, чемпионат такой по кислотно-основным равновесиям – никаких предсказаний (ставки делать можно), одни игры (измерения). Дело на самом деле еще хуже, ведь в другом растворителе равновесия были бы другие, и нам пришлось бы еще для каждого важного ратворителя все перемерить. А кислот не десять, и даже не сто. И оснований тоже немало. Тупик.
К счастью, это не так. Давайте попробуем из водных уравнений констант сохранить одну важную вещь – представим, что и в неводном растворе константа равновесия реакции кислота-основание равна отношению констант кислотности кислоты и сопряженной кислоты основания. Но мы их не знаем! Их не знаем, а отношение их знаем, потому что можем его измерить. Это работает потому, что так устроено химическое равновесие (и нехимическое тоже, но это не наши проблемы). Но пока не ясно, в чем облегчение – все равно измерять придется конкретные константы равновесия для каждой пары.
Представьте себе, что у вас две кислоты и одно основание. И вы измеряете константу равновесия реакции первой кислоты с основанием, и второй кислоты с основанием. И находите, что вторая константа равновесия, например, в десять раз больше первой. Значит ли это, что вторая кислота в десять раз сильнее первой? Да, значит, и это легко проверить. Находим второе основание и измеряем константы равновесия с обеими кислотами. И, да – вторая константа опять будет в 10 раз больше первой (с учетом точности эксперимента). И теперь у нас 4 константы, и мы можем сравнить уже первую кислоту с первым и вторым основанием и убедиться, что отношение будет таким же, как у второй кислоты с первым и вторым основанием – мы определили разницу в основности (или кислотности сопряженной кислоты) оснований. А это возможно только, если в неводной среде, как и в водной, есть вполне определенные константы кислотности и основности. Но – мы по-прежнему их не знаем, знаем только их отношения.
Шкалы констант кислотности и основности в неводных средах
Итак, мы выяснили, что константы кислотности в каком-то важном растворителе можно свести в одну таблицу или шкалу – от слабых кислот к сильным. Про основность давайте уже примем, что константы основности легко и удобно сводятся к константам кислотности, так что можно и даже нужно располагать единой шкалой констант кислотности для всех кислот и оснований, в которой основания представлены константой кислотности сопряженных кислот. Но эта шкала пока не привязана к какому-то стандарту и является строго относительной. Это хорошо или плохо?
Но вообще-то это именно то, что реально нужно – ведь нам требуется только предсказывать константы равновесий между кислотами и основаниями. И если мы можем расположить все интересующие нас кислоты и основания, и мы можем все это хозяйство расположить в виде множества относительных кислотностей, то деля одно на другое для любой пары из этого множества, мы сможем получать вполне корректные константы равновесий. И чисел у нас для примерного множества из 10+10 будет не сто, и даже не 20, как в воде, а всего 19. И одно произвольное число, которое можно без ущерба для математики принять за единицу (если непонятно, откуда это, сведите множество к минимуму – к двум кислотам, и пусть, например, одна кислота в 10 раз сильнее другой – тогда для характеристики этих двух кислот пойдут числа 1 и 10, или 2 и 20, или 1000 и 10000, или 3.14 и 31.4 и т.п.). Это типичная проблема для экспериментальных естественных наук – это называется или выбор единицы измерения или калибровка.
Вот мы приехали к очень странному результату. В неводной среде можно получить набор чисел, которые в расчетах констант равновесий реакций кислота-основание играют роль констант кислотности, но сами величины эти могут быть вполне произвольны, если сохраняются их отношения. Раз числа произвольны, это не настоящие константы кислотности, а относительные. Но поскольку мы все равно не можем понять, что такое настоящие константы кислотности в неводной среде и как нужно корректно записать закон действия масс (уравнение равновесия) для одной кислоты или основания в неводном растворе, то придется смириться с такой несправедливостью. И еще придется смириться с тем, что в разных растворителях шкалы будут разными (это еще не самая плохая новость – настоящий кошмар впереди).
То, что шкалы в разных растворителях окажутся разными для практических целей не имеет большого значения, но с эстетической точки зрения страшно раздражает. Значения это не имеет, потому что числа из разных шкал никогда не встретятся в одном расчете – невозможно перенести протон не только от кислоты к основанию, но параллельно еще из одного растворителя в другой. Мысленно такой эксперимент возможен, но реально нет. Но вполне возможно изучить реакции кислот и оснований в смеси растворителей. И тогда мы либо должны будем еще и перемерить все шкалы в различных смесях, что уже совсем похоже на сизифов труд, причем сам Сизиф, если бы ему предложили альтернативу катать камень или измерять константы, почти наверняка предпочел бы продолжать свою античную забаву. Либо мы должны как-то интерполировать между двумя шкалами в чистых растворителях, а тогда все же желательно их как-то разумно заякорить (откалибровать, задать масштаб) и не давать им свободно елозить по числовой оси.
Обычно все же вместо того, чтобы мириться, пытаются сделать одну немного неточную, но полезную вещь. Для того, чтобы ее сделать, задают вопрос, а почему константы в разных растворителях разные. Видимо, виновата сольватация, взаимодействие кислот и оснований из равновесий с растворителем. А можно ли ее, энергию сольватации, найти (измерить, рассчитать, предсказать на основании исследования внутренностей жертвенных животных, и т.п.)? Ответ: попробовать можно, и многие пробовали и до сих пор пробуют, но это сложно и ненадежно, если делать это для каждой молекулы или иона, задействованных в равновесиях.
Тогда можно сделать проще – найти такие кислоты или основания, у которых сольватация относительно слаба и сводится только к самым простым свойствам сред. Здесь уже все проще, потому что хорошо известно, что сольватируется сильно, а что слабо. Сильно сольватируются любые частицы с концентрированным зарядом и с хорошими возможностями для химического взаимодействия (наличие неподеленных пар, пустых орбиталей, доноров или акцепторов водородной связи и т.п.). Если всего этого нет, или хотя бы мало, то можно считать, что растворитель относительно слабо взаимодействует с такими молекулами и ионами, и можно или совсем этим пренебречь, или ввести небольшую поправку, например, на полярность среды.
Для каждой кислоты или основания мы имеем две частицы, одна из них не заряжена, а другая является катионом или анионом. Анионы почти всегда сольватируются сильно, потому что отрицательный заряд почти всегда связан с неподеленной парой и акцептором водородной связи. А вот катионы бывают очень слабо взаимодействующие с растворителем: это, например, большие катионы аммония R3NH+. Заряд в них не связан с реакционноспособным центром, и если R довольно большие группы, то этот заряд еще и зарыт внутри довольно большой частицы, там же зарыт и атом водорода, могущий участвовать в водородной связи.
Стоит нам найти хотя бы одну такую кислоту или основание, как мы можем сказать, что ее константа кислотности приблизительно одинакова в разных средах, а тогда почему и не в воде, и если так, то почему бы нам не выбрать такую кислоту или основание за якорь всей системы констант. Взять ее за точку отсчета шкалы, но вместо единицы использовать значение константы кислотности в воде. Тогда мы получим нечто очень привлекательное: табличку чисел, принимаемых нами за константы кислотности в каком-то неводном растворителе, причем некоторые из этих чисел будут очень близки к числам из другой таблицы, таблицы констант кислотности в воде, а некоторые будут сильно, а иногда и очень сильно, на многие порядки, различаться. И мы узнаем, что кислотность или основность в неводном растворителе для некоторых кислот и оснований может быть намного – в разы, на порядки – больше чем в воде. Это очень важное знание и мы будем часто им пользоваться.
Можно ли в органическом растворителе использовать индикаторные бумажки?
Можно, но смысла в этой процедуре немного – вы ничего не измерите, и в лучшем случае просто узнаете, что в растворе есть кислота (если нет – не узнаете). В органическом растворителе нет такого показателя среды как pH, и оценить его индикатором нельзя. При этом цвет они менять будут, особенно в присутствии кислоты. Причина этого проста – изменение цвета индикаторов в кислой среде почти всегда связано с протонированием молекул индикатора. Индикаторы – почти всегда основания с разной окраской кислотной и основной формы. Они будут протонироваться кислотами в любом растворителе, если кислотность кислоты достаточна (индикаторы – всегда слабые основания). Поэтому, если кислота достаточно сильная, вы увидите изменение окраски бумажки, если слабая, то, скорее всего, не увидите. Насколько сильно изменится цвет сказать невозможно, и степень изменения окраски не будет иметь никакого отношения к константе кислотности. Это будет нечто случайное, особенно, если бумажка влажная, и в порах бумаги возникнет случайная Аррениусова кислотность. С основностью все будет еще менее предсказуемо, потому что индикаторы подбирают именно для водных растворов, и химия изменения цвета в основной части значений pH не всегда связаны с простым отъемом протона, а бывает, что требует взаимодействия с настоящим гидроксид-ионом, которого в органической среде нет. И снова возможны фокусы, если бумажка влажная. Результат будет довольно случаен, и с тем же успехом можно бросать монетку или игральную кость, типа, выпадет орел или шестерка – есть кислота, не выпадет, нет.
Не будем дивить людей и пихать индикаторные бумажки в неводные растворы, даже если нас к этому толкает какая-нибудь непродуманная методика.
Почему мы так любим использовать pK, когда речь идет о константах кислотности и основности
pK – это десятичный логарифм с обратным знаком, точно то же самое, что подразумевает маленькая буква p в pH. Очевидно, что это просто удобно. Константы равновесий, в том числе константы кислотности и основности – это что-то на десять в минус какой-то степени, неуклюжая величина, которую просто так не прочитаешь, и со слуха не воспроизведешь. А pK – это просто число, обычно небольшое и положительное, меньше 50, это легко запомнить, считать со слуха, да и сравнивать тоже удобно, только нужно не забывать, что
- для кислот чем больше pKa, тем меньше кислотность
- для оснований, если основность выражена константой кислотности сопряженной кислоты, а это так и есть в большинстве случаев, то чем эта величина больше, тем больше основность
- для оснований, тем не менее, нужно бдительно следить, чтобы вам не подсунули именно константу основности и ее pKb, и если вам не повезло и это так, то чем эта величина больше, тем меньше основность. Подлость заключается в том, что основность, выраженную кислотностью сопряженной кислоты, иногда неряшливо и ошибочно называют именно константой основности и pKb.
Но есть и другая и существенно более важная причина, чем удобство (хотя что может быть важнее, чем удобство!). Логарифм от константы равновесия имеет размерность энергии, и действительно это ни что иное как свободная энергия, точнее ее изменение, связанное с реакцией, изображающей равновесие. Взяв эту величину с обратным знаком, мы просто и получаем оценку изменения свободной энергии ΔG для прямой реакции в равновесии. Напомню, что положительная величина ΔG это плохо, это значит, что реакция вперед идти не хочет, и чем больше эта величина, тем больше не хочет. Если в равновесии прямая реакция идти не хочет, это непосредственно значит, что хочет обратная (для нее ΔG имеет ту же величину, но обратный знак). Когда мы сравниваем pK двух кислот или двух оснований, мы просто непосредственно получаем ответ на то, кто у кого может оторвать протон, и насколько легко пойдет этот процесс. Только нужно не запутаться в том, что из чего нужно вычитать. Займемся этим чуть позже.
В каких единицах энергии получаются величины, полученные из pK? Строго говоря, это не важно, потому что мы всегда имеем дело не с самими величинами, а с их сравнением – больше-меньше. Но при желании можно и строго на этот вопрос ответить, потому что это просто – нужно просто помножить на RT, а эта величина при комнатной температуре приблизительно равна 0.6 ккал/моль. То есть разница pK в 10 единиц приблизительно соответствует разнице свободных энергий в 6 ккал/моль. Если вы привыкли к другим единицам энергии, просто найдите значение RT в этих единицах и пользуйтесь им.
“Плохие” и “хороший” растворители
А теперь плохое и хорошее известие в одной фразе: в каждом растворителе должна получиться своя шкала кислотности/основности (плохое известие – шкал много, запутаемся, пропадем), но в реальности существует (измерено, опубликовано) только несколько шкал, а реально используется только одна (хорошее известие – мы спасены!). Точнее одна, кроме уже хорошо известной водной.
Причина такой скромности очень простая и она связана не с трудоемкостью – будьте уверены, что нашлись бы желающие измерить хоть тысячу растворителей, – а в скудности того набора растворителей, который существует и доступен для использования. И большинство из существующих растворителей вообще непригодны для корректных измерений равновесий с участием заряженных частиц. Большинство органических растворителей малополярны (что это точно означает, рассмотрим немного позже), и в таких растворителях ионы очень сильно взаимодействуют друг с другом, образуя агрегаты переменного состава. Равновесия с участием таких сложных частиц накладываются на собственно кислотно-основное равновесие, и непредсказуемо изменяют измеряемые константы равновесий. Получаются очень сложные зависимости от концентраций, из которых сложно или даже вообще невозможно извлечь константы кислотности и основности.
Когда мы обсуждали константы кислотности в неводных растворителях, проделывали мысленный эксперимент с двумя кислотами и двумя основаниями, и утверждали, что если в реакциях с первым основанием одна кислота оказалась в столько-то раз сильнее второй, то и по отношению к второму основанию отношение будет тем же, приблизительно. Именно из этого мы вывели возможность измерения шкалы кислотности и использовании ее для оценки констант равновесий в реакциях кислот и оснований. Но это возможно только, если катион и анион взаимодействуют относительно слабо, и тогда мы можем разделить кислотную и основную пары. В воде именно так – там катионы и анионы разделены и свободны. А в малополярной органике это не так, и каждая пара анион-катион взаимодействует сильно и фактически непредсказуемо. В том мысленном эксперименте отношение кислотностей в реакциях с разными основаниями будет разным – и сильно. И какое выбрать?
Поэтому и нет у нас под рукой шкал кислотности в толуоле, этаноле, эфире, ацетоне и т.п. Даже в ТГФ нет хорошей шкалы, а ведь это очень скверно, потому что ТГФ очень часто используют для кислотно-основных реакций с так называемыми CH-кислотами, а это просто одна из самых важных реакций в органике.
В этом смысле мы можем назвать большинство органических растворителей “плохими”. Это не значит, что в них не идут реакции кислот и оснований. Но это значит, что мы не сможем даже грубо оценить константы равновесий таких реакций. А нам это может понадобиться? Это сложный вопрос и мы к нему еще вернемся. Пока скромно заметим, что мы бы не отказались, если бы это было возможно. Это упростило бы многие проблемы.
Но органика все же не безнадежна. Среди органических жидкостей есть полярные среды, в которых ионы чувствуют себя более-менее свободно. Не совсем так, как в воде, но неплохо. Самая знаменитая из таких жидкостей называется диметилсульфоксид (ДМСО). Эта жидкость обладает высокой полярностью, сравнимой с полярность воды (пока без подробностей, но это так), она очень слабо взаимодействует с анионами, и довольно сильно – с катионами, особенно такими, которые имеют валентные возможности для взаимодействия (катионами металлов, небольшими протонированными частицами). В этом растворителе получается хорошо воспроизводимая шкала кислотностей (или основностей, выраженных как кислотность сопряженных кислот).
Очень большое количество таких данных измерил и собрал из других работ канадский ученый Ф.Бордвелл. Таблицы доступны в сети. ДМСО в этом смысле – хороший растворитель.
Как же это получается – плохих много, а хороший один? Не будем драматизировать – растворителей, похожих на ДМСО довольно много, но они похожи друг на друга, и шкала кислотностей в любом таком растворителе будет очень похожа на шкалу в ДМСО, поэтому большого смысла измерять и публиковать ее нет.
Что такое “сильная кислота”?
В воде “сильной кислотой” мы называем кислоту, которая полностью диссоциирована в разбавленных растворах. Следовательно у сильных кислот нет констант кислотности, просто потому что в знаменателе стоит концентрация недиссоциированной формы, а она равна нулю, и константа кислотности стремится к бесконечности, а значит не имеет смысла. Если вспомнить, что в воде кислота передает протон воде, то становится понятно, что сильной кислотой в воде является кислота, способная количественно протонировать воду (сколько положили кислоты, столько и будет ионов гидроксония).
Из всего этого следует один чрезвычайно важный вывод:
в воде все сильные кислоты одинаковы
и отличаются только молярной массой. Если кислота сильная, то это все, что про нее можно сказать. Не может быть более сильной сильной кислоты и менее сильной сильной кислоты. В воде нет разницы между серной, хлорной, фторсульфоновой, тетрафторборной, толуолсульфоновой и т.п. кислотами, но нелишне помнить, что с случае многоосновных кислот типа серной, только первая ступень диссоциации соответствует высокому званию “сильной кислоты”. Раствор сильной кислоты легко характеризовать pH раствора, который очень легко прикинуть. Корректности ради не забудем, что речь идет о разбавленных растворах. В концентрированных растворах сильные кислоты ведут себя так, как будто мы имеем дело с неводной средой.
В неводных средах все кислоты разные, хотя растворитель и накладывает огрничение на силу кислоты, о чем скажем ниже. Отлично может быть более сильная сильная кислота, и еще более сильная сильная кислота, и совсем сильная сильная кислота, и чрезвычайно сильная сильная кислота, и самая-наисамая сильная кислота, и лет сорок назад был даже чемпионат по силе кислот самых сильных кислот или суперкислот, который был выигран чрезвычайно предприимчивым американским ученым венгерского происхождения Джорджем Олой (нобелевский лауреат 1995 года) с Волшебной кислотой (Magic acid). С тех пор ажиотаж как-то спал, и этот титул время от времени кто-нибудь оспаривает, но на это уже никто не обращает внимания.
Как же узнать, какая кислота сильнее. Да очень просто – мы же уже разобрались, что в неводных средах кислота передает протон основанию, и степень переноса протона можно узнать, если знать константы кислотности (pK). А если не знать, то можно поступить совсем просто. Представьте себе, что у вас есть набор оснований (в прямом смысле – коробочка такая, в которой лежат пузыречки с основаниями), и все эти основания кто-то заботливо расположил по убыванию основности: слева лежат более сильные основания, справа – более слабые. И еще мы для каждого основания можем легко определить, запротонировалось ли оно, и насколько – имеем какой-нибудь удобный спектроскопический метод, который позволяет быстро различить основание и его сопряженную кислоту, и измерить их относительные концентрации. Готовый набор из 17 оснований, перекрывающий огромный диапазон основности, предложил еще в 1932 году основоположник теории органической химии Луис Гаммет, и этот набор, уже готовый, можно прямо купить в магазине (номер по каталогу Сигма-Олдрич-Мерк 14,383-9). А можно и самостоятельно составить, порывшись в лаборатории на полках, но придется по литературе разузнать константы основности и характеристики основной и кислотной форм. Такие наборы принято называть наборами индикаторов, и вполне удачно, так как они выполняют ту же роль, что и наборы красителей, изменяющих цвет для определения pH в водном растворе.
Итак, берем первую кислоту и добавляем одинаковое количество в отсортированный по убыванию основности ряд пузырьков с растворами индикаторов. Замечаем, что в пузырьках слева происходит протонирование, а пузырьках справа нет. Где-то в середине будет ряд растворов с частичным протонированием, и мы можем определить степень протонирования и увидеть что она падает слева направо. Берем вторую кислоту и проделываем с ней то же самое. Если увидим, что она протонирует больше индикаторов и заезжает дальше направо, то скажем, что эта кислота сильнее первой. А если наоборот – меньше индикаторов и останавливается ближе к левому концу ряда пробирок с растворами индикаторв – то слабее. И так далее со всеми кислотами, которые у нас есть. Если повезет, то найдем такие кислоты, которые протонируют почти все индикаторы – и такие кислоты будут самыми сильными из тех, что нам удалось найти. Почти наверняка нам не удастся найти кислот, которые протонируют все индикаторы, особенно если у нас есть готовый набор Гаммета – с правой стороны там такие слабые основания, что они не протонируются в растворе концентрированной серной кислоты. Но есть на свете кислоты, которые и их протонируют – это суперкислоты. Мы еще к этому вернемся, поэтому здесь без подробностей – только общая идея, как определить относительную силу сильных кислот.
Делаем выводы:
- в воде понятие “сильная кислота” качественное, все сильные кислоты по кислотности одинаковы, а между сильными и слабыми кислотами существует принципиальная разница (например, в том, что у первых нет константы кислотности, а у вторых есть).
- в неводных растворах сильная кислота – понятие количественное, все сильные кислоты различаются по кислотности, и нет принципиальной разницы между сильными кислотами и слабыми – все кислоты можно выстроить в ряд по протонирующей способности по отношению к основаниям разной основности. У любой кислоты, как бы сильна она не была, есть значение константы кислотности (pKa), и у очень сильных кислот это отрицательные величины.
Оценку кислотности кислот по набору индикаторов можно сделать в любом растворителе. Но есть одна важная характеристика растворителя, определяющая, какую максимальную кислотность можно в нем получить. Да, растворители ограничивают диапазон возможной кислотности сверху, и то что было сказано раньше – в неводных растворителях все кислоты разные, вообще говоря, неверно. Многие растворители являются основаниями (строго говоря, почти все растворители являются основаниями, а еще строже – если найдете растворитель, который не является основанием, сообщите мне, и чур, жидкие гелий, неон, аргон, криптон и ксенон не предлагать). И у каждого растворителя есть собственная основность, у одних большая, у других меньшая. И тогда может получится, а точнее обязательно получится так, что некоторые из индикаторов будут иметь меньшую основность чем молекулы растворителя, и протонироваться будет не индикатор, а растворитель (не забывайте еще, что концентрация растворителя в растворе всегда больше, сем концентрация растворенного вещества, поэтому даже при сравнимой основности растворителя и индикатора протонироваться будет в основном растворитель). Понятно, что все еще более сильные кислоты протонировать будут только растворитель, и мы потеряем возможность судить о том, что они в этом растворителе сильнее. Собственная основность растворителя устанавливает потолок достижимой кислотности. Мы ведь именно это уже видели в воде – собственная основность воды довольно велика. А если мы возьмем в качестве растворителя, например, эфир или ТГФ, которые обладают даже большей основностью чем вода, получим почти то же самое, что в воде – мы не сможем различить кислот, более сильных чем, условно, HCl. А если возьмем в качестве растворителя такое неслабое основание как триэтиламин, то не сможем различить уксусную кислоту от серной. И наоборот, если взять более слабое основание, например, ацетон, то диапазон кислотности вырастет, хотя и не очень сильно.
Теперь мы, наверное, легко поймем, что с суперкислотами в реальности проблем очень много – их нельзя различить в подавляющем большинстве растворителей, потому что они их протонируют, и протон не доходит до индикаторов со слабоосновного конца набора. И для того чтобы искать самую сильную кислоту – Волшебную кислоту – Джорджу Оле пришлось сначала придумать очень экзотический и чрезвычайно слабоосновный растворитель, потому что во всех обычных растворителях разницу в кислотности суперкислот установить невозможно.
Что такое “сильное основание”?
К сильным основаниям применимы почти те же рассуждения, что и к сильным кислотам, но с некоторыми важными изменениями.
В воде ситуация очень похожа. Начиная с некоторого уровня основности мы теряем возможность различать основания по силе. Вода – слабая кислота, и любое сильное основание количественно ее депротонирует, возвращая в раствор одно и то же – гидроксид-ион. Поэтому самое сильное основание в воде – это и есть сам гидроксид, который можно просто взять в виде гидроксида щелочного металла, или просто щелочи.
Сильное основание в воде – это щелочь, только щелочь, и ничего кроме щелочи.
Совершенно бессмысленно в водных растворах использовать более сильные основания, например, алкоксиды или гидриды – получите эквивалентное количество щелочи, которая заведомо дешевле и доступнее.
В неводных растворителях диапазон основностей гораздо шире. Так как мы договорились характеризовать основность через кислотность сопряженной кислоты, получим. что задача расположения оснований по силе ничем не отличается от задачи расположения сопряженных кислот по кислотности. Чем слабее сопряженная кислота, тем сильнее основание. И наоборот. Как измеряют pKa для очень сильных оснований обсудим как-нибудь отдельно. В принципе, это неважно.
Важно понимать, что растворитель так же ограничивает силу основания, как и силу кислоты. Растворитель может оказаться кислотой, и тогда все основания, превосходящие по силе сопряженное основание растворителя, будут его депротонировать, и единственным основанием в таких растворах и окажется сопряженное основание растворителя. Но картина здесь намного более приятная, чем в случае силы сильных кислот. Найти растворитель, который бы не являлся основанием, почти невозможно, но далеко не все растворители являются кислотами. Чтобы быть кислотой растворитель должен иметь протоны, которые можно оторвать достаточно сильным основанием. Понятно, что наличие OH, NH, SH в составе молекул растворителя заведомо делает их кислотами, но и многие связи CH также уязвимы.
Поэтому, например, в растворе метанола самым сильным основанием является метоксид, и бесполезно растворять метаноле трет-бутилат калия или амид натрия – будет тот же метоксид, заведомо более слабое основание чем любое из взятых.
А, например, в растворе жидкого аммиака отлично можно использовать метоксид и даже трет-бутилат, и это будут разные основания. Самым сильным основанием будет амид NH2–. Бесполезно растворять в жидком аммиаке заведомо более сильное чем амид основание, например, бутиллитий.
Такие вещи стоит иметь в виду. Иногда ситуация в том или ином растворителе совсем не очевидна. Тогда нужно искать pKa и оценивать основность. Вот, например, уже знакомый нам диметилсульфоксид (ДМСО) – почему бы не растворить в нем амид натрия. Но оказывается, ДМСО это слабая кислота с pKa = 35, а основность амид-аниона, выражаемая через кислотность аммиака – pKa = 38! Амид – более сильное основание, чем сопряженное основание ДМСО (его иногда называют димсил-анионом), и это значит, что растворяя амид натрия в ДМСО, мы больше не имеем амид, а имеем немного более слабое основание. Во многих случаях это непринципиально, но иногда лишние 3 единицы pK очень важны. А вот трет-бутилат калия в ДМСО растворить очень даже рекомендуется – получите раствор очень сильного основания (pKa трет-бутанола в ДМСО около 31).
Обратите внимание, что ограничение, накладываемое собственно кислотностью растворителя, не означает, что в таком растворителе не может быть оснований с pK большим, чем у растворителя. Не забывайте, что в константы равновесия входят еще и концентрации, а современные методы анализа позволяют определять очень малые концентрации. Если вы посмотрите на таблицу pKa в ДМСО, то найдете там основания с величинами больше 35. Это совсем другая проблема – как измерить или оценить такое pKa в таком растворителе. Это возможно, хотя чем больше разница, тем меньше надежность такой оценки и меньше точность. Для практических целей в этом мало смысла, потому что какое бы ни было pKa, реальная основность, достижимая в растворе, ограничена свойствами растворителя. Мы еще вернемся к этой важной теме, когда будем, например, выяснять, каким образом заведомо более слабые основания умудряются участвовать в реакциях, требующих депротонирования очень слабых кислот и образования более сильных сопряженных оснований.
