Квантовый выход (Φ) излучательного процесса – величина, равная отношению количества раз, когда конкретное событие происходит, к количеству поглощенных квантов возбуждающего излучения.

Приложения[править | править код]
Люминесцентная спектроскопия[править | править код]
Квантовый выход люминесценции определяется как отношение количества испускаемых фотонов к количеству поглощенных фотонов. [1]
 ,
,
где Nem — количество излученных фотонов, а Nabs — количество поглощенных фотонов.
Квантовый выход люминесценции измеряется по шкале от 0 до 1, но часто выражается в процентах. Квантовый выход 1 (100%) описывает процесс, в котором каждый поглощенный фотон приводит к испускаемому фотону. Многие современные комплексы, в частности органические люминофоры на основе ионов лантаноидов, имеют теоретически квантовый выход на уровне 99%, однако реальный квантовый выход из-за различных побочных неизлучательных процессов значительно ниже.
Квантовый выход определяется долей люминофоров в возбужденном состоянии, которые прорелаксируют в основное состояние через люминесценцию:

где Φf — квантовый выход люминесценции, kf — константа скорости излучательной релаксации (люминесценции), knr– константа скорости всех процессов безызлучательной релаксации. Безызлучательные процессы представляют собой механизмы релаксации из возбужденного состояния, отличные от испускания фотонов, которые включают: Фёрстеровский перенос энергии, внутренняя конверсия и интеркомбинационная конверсия(ISC). Таким образом, на квантовый выход люминесценции влияет изменение скорости любого безызлучательного процесса. Квантовый выход может быть близок к единице, если скорость безызлучательного распада намного меньше скорости излучательного распада, т. е.  [1]
[1]
Фотохимические реакции[править | править код]
Квантовый выход фотохимической реакции описывает количество молекул, подвергающихся фотохимическому событию, на один поглощенный фотон:

Квантовый выход больше 1 возможен для фотоиндуцированных или радиационно-индуцированных цепных реакций, в которых один фотон может вызвать длинную цепочку превращений. Одним из примеров является реакция водорода с хлором, в которой может образоваться до 106 молекул хлороводорода на квант поглощенного синего света[2]. Тут необходимо отметить, что делается допущение о незамкнутости системы, так как выделяя отдельно систему из фотона и поглощающей его частицы, мы не можем получить квантовый выход более 1.
Квантовый выход фотоэффекта[править | править код]
Важной количественной характеристикой фотоэффекта является квантовый выход Y — число эмитированных электронов в расчёте на один фотон, падающий на поверхность тела. Величина Y определяется свойствами вещества, состоянием его поверхности и энергией фотонов.
Квантовый выход фотоэффекта из металлов в видимой и ближней УФ-областях Y < 0,001 электрон/фотон. Это связано, прежде всего, с малой глубиной выхода фотоэлектронов, которая значительно меньше глубины поглощения света в металле. Большинство фотоэлектронов рассеивает свою энергию до подхода к поверхности и теряет возможность выйти в вакуум. При энергии фотонов вблизи порога фотоэффекта большинство фотоэлектронов возбуждается ниже уровня вакуума и не даёт вклада в фотоэмиссионный ток. Кроме того, коэффициент отражения в видимой и ближней УФ-областях велик и лишь малая часть излучения поглощается в металле. Эти ограничения частично снимаются в дальней УФ-области спектра, где Y достигает величины 0,01 электрон/фотон при энергии фотонов E > 10 эВ.
Фотосинтез[править | править код]
Квантовый выход используется при моделировании фотосинтеза[3]:

Измерение квантового выхода фотолюминесценции[править | править код]

Принцип измерения квантового выхода так же прост, как сложна его реализация. Существует два основных принципа измерения квантового выхода: абсолютный, фактически использующий определение квантового выхода как отношения числа излученных и поглощенных фотонов, и относительный, в котором исследуемый образец сравнивается с известным стандартом.
Измерение абсолютного квантового выхода проводится с использованием интегрирующей сферы, в которую помещается образец и к которой проводят волноводы, ведущие к источнику возбуждения и к детектору. Интегрирующая сфера обеспечивает попадания всего отраженного и излученного света на детектор.
Принцип измерения чрезвычайно прост. В одинаковых условиях проводится измерение спектра люминесценции исследуемого образца (Ec), спектра люминесценции пустой кюветы (Ea), спектра релеевского рассеяния образца (Lc) и спектра релеевского рассеяния пустой кюветы (La). Поскольку интенсивность люминесценции образца соответствует (Ec-Ea), а поглощения – (La-Lc), квантовый выход можно выразить как
 [4].
[4].
При относительном измерении квантового выхода квантовый выход изучаемого соединения (Qx) определяется по формуле
 ,
,
где Qs — квантовый выход образца сравнения, E – площадь под спектром люминесценции, A(λ)— поглощение на длине волны возбуждения, I(λ) – интенсивность возбуждающего пучка на длине волны возбуждения, n – коэффициент преломления. От сомножителя  обычно можно избавиться, если проводить измерения образца сравнения и исследуемого образца в одинаковых условиях при одной длине волны возбуждения. Кроме того, если в качестве длины волны возбуждения выбирать длину волны, при которой спектры поглощения образца сравнения и исследуемого образца пересекаются, то сомножитель
обычно можно избавиться, если проводить измерения образца сравнения и исследуемого образца в одинаковых условиях при одной длине волны возбуждения. Кроме того, если в качестве длины волны возбуждения выбирать длину волны, при которой спектры поглощения образца сравнения и исследуемого образца пересекаются, то сомножитель  становится равен 1, и выражение упрощается до
становится равен 1, и выражение упрощается до
 .
.
Для большей достоверности полученных результатов рекомендуется проводить измерения при возбуждении несколькими разными длинами волн.
Относительный квантовый выход флуоресценции измеряют путем сравнения со стандартом известного квантового выхода. Соль хинина сульфат хинина в растворе серной кислоты считалась наиболее распространенным стандартом флуоресценции[5], однако недавнее исследование показало, что квантовый выход флуоресценции этого раствора сильно зависит от температуры и больше не должен использовать как стандартный раствор. Хинин в 0,1 М хлорной кислоте (Φ=0,60) не показывает зависимости от температуры до 45°С, поэтому его можно рассматривать как надежное стандартное решение[6].
| Люминофор | Растворитель | λex ,nm | Φ |
|---|---|---|---|
| Хинин | 0,1 М 
|
347,5 | 0,60 ± 0,02 |
| флуоресцеин | 0,1 М 
|
496 | 0,95 ± 0,03 |
| Триптофан | Вода | 280 | 0,13 ± 0,01 |
| Родамин 6G | Этиловый спирт | 488 | 0,94 |
Закон Вавилова[править | править код]
Формулировка закона:
Квантовый выход постоянен при изменении в широких пределах длины волны возбуждающего света в стоксовой области и падает, если длина волны возбуждающего света лежит в антистоксовой (длинноволновой) области спектральной полосы поглощения.
В соответствии с постоянством квантового выхода энергетический выход растёт с увеличением длины волны возбуждающего света и падает в антистоксовой области.
Закон справедлив только при изменении длины волны возбуждающего света в пределах одной электронной полосы поглощения. Если при фотовозбуждении молекулы переходят в различные электронные состояния, то квантовый выход может меняться и закон не будет выполняться. Закону подчиняется люминесценция твёрдых и жидких растворов люминесцирующих веществ, молекулярных кристаллов, кристаллофосфоров при поглощении света в активаторе.
Падение квантового и энергетического выхода при возбуждении светом с длиной волны, лежащей в антистоксовой области, связано с уменьшением в этой области вероятности электронного перехода на возбуждённый уровень. Неселективное и невозбуждающее люминесценцию поглощение примесями или основным веществом оказывается больше возбуждающего люминесценцию, это приводит к уменьшению доли возбуждающих люминесценцию квантов из всех поглощённых, т. е. к падению выхода люминесценции[7].
Примечания[править | править код]
- ↑ 1 2 Lakowicz, Joseph R. Principles of Fluorescence Spectroscopy (Kluwer Academic / Plenum Publishers 1999) p.10. ISBN 978-0-387-31278-1
- ↑ Keith J. Laidler. Chemical kinetics. — 3rd ed. — New York: Harper & Row, 1987. — xi, 531 pages с. — ISBN 0-06-043862-2, 978-0-06-043862-3.
- ↑ John B. Skillman. Quantum yield variation across the three pathways of photosynthesis: not yet out of the dark // Journal of Experimental Botany. — 2008. — Т. 59, вып. 7. — С. 1647–1661. — ISSN 1460-2431. — doi:10.1093/jxb/ern029. Архивировано 9 марта 2022 года.
- ↑ Уточникова Валентина Владимировна. [http://www.inorg.chem.msu.ru/lcc/new/pages/files/lum_metod.pdf методическая разработка к спецкурсу
ЛЮМИНЕСЦЕНЦИЯ органических соединений] / Рекомендовано Методической комиссией Химического факультета и Факультета наук о материалах МГУ в качестве учебного пособия для студентов старших курсов в 2014 г. — МГУ им М.В. Ломоносова, 2014. — С. 33. Архивировано 11 июля 2019 года. - ↑ Standards for Photoluminescence Quantum Yield Measurements in Solution (IUPAC Technical Report) // Chemistry International — Newsmagazine for IUPAC. — 2011-01. — Т. 33, вып. 6. — ISSN 0193-6484 1365-2192, 0193-6484. — doi:10.1515/ci.2011.33.6.34c.
- ↑ Goodbye to Quinine in Sulfuric Acid Solutions as a Fluorescence Quantum Yield Standard. dx.doi.org. Дата обращения: 14 марта 2022.
- ↑ Sergei I. Vavilov. Выход флуоресценции растворов красителей в зависимости от длины волны возбуждающего света. II // Uspekhi Fizicheskih Nauk. — 1967-10. — Т. 93, вып. 10. — С. 315–320. — ISSN 1996-6652 0042-1294, 1996-6652. — doi:10.3367/ufnr.0093.196710f.0315.
Лекция 36
Фотохимические реакции. Основные законы фотохимии. Квантовый выход. Общее уравнение скорости фотохимической реакции. Типы фотохимических процессов.
ОСНОВНЫЕ ЗАКОНЫ ФОТОХИМИИ. КВАНТОВЫЙ ВЫХОД.
Фотохимией называется раздел физической химии, изучающий закономерности протекания химических процессов, обусловленных действием света.
Основной процесс, проходящий под действием света, – фотодиссоциация, или возбуждение молекул.
Гротгус в России (1817) и Дрейпер в США (1839) независимо друг от друга сформулировали закон, согласно которому химически активны лишь те лучи, которые поглощаются реакционной смесью. Этот закон очевиден и не имеет исключений. Закон Гротгуса-Дрейпера непосредственно связывает химическое действие света с его поглощением веществом.
Ламберт (1760) установил, что ослабление интенсивности dI света, прошедшего через слой толщиной dl, прямо пропорционально толщине слоя и интенсивности падающего света I, а Бер (1853) показал, что поглощение тонким слоем прямо пропорционально числу частиц (молекул) или их концентрации в слое. Объединенный закон Ламберта-Бера можно записать в форме :
I = Ioe–knl (1)
Io – интенсивность светового потока до прохождения поглощающего слоя;
Рекомендуемые материалы
I – то же после поглощения в слое толщиной l;
n – число поглощающих свет молекул в 1 см3;
k – множитель пропорциональности, называемый молекулярным коэффициентом поглощения.
Вант-Гофф показал (1904), что количество химически измененного вещества прямо пропорционально количеству поглощенной веществом световой энергии. Количество энергии Q, поглощенной в единицу времени, может быть найдено из закона Ламберта-Бера :
Q = Io – I = Io (1 – e-knl) (2)
Тогда скорость фотохимической реакции  пропорциональна количеству энергии, поглощенной веществом в единицу времени :
пропорциональна количеству энергии, поглощенной веществом в единицу времени :
 –
–  = K Io (1 – e–knl) (3)
= K Io (1 – e–knl) (3)
К – множитель пропорциональности.
Наиболее интересным и важным законом, позволившим разобраться в механизме фотохимических реакций, является закон фотохимической эквивалентности Штарка-Эйнштейна (1912), который гласит, что каждому поглощенному кванту излучения hn соответствует одна измененная молекула.
Число квантов, поглощенных в единицу времени, равно na = Q/hn. Следовательно, изменению под действием света должны подвергнуться np = Q/hn молекул.
Опыт показывает, что во многих случаях число фотохимически прореагировавших молекул не равно числу поглощенных квантов. Поэтому для характеристики фотохимических реакций было введено понятие квантового выхода g. Квантовым выходом называется отношение числа прореагировавших молекул к числу поглощенных квантов :
g =  =
=  (4)
(4)
где np – число прореагировавших молекул.
Скорость химической реакции
–  =
=  = g
= g  = g
= g  (5)
(5)
Подставив в уравнение (5) выражение (2) , получим
– = g  (1 – е–knl) (6)
(1 – е–knl) (6)
Это наиболее общее выражение для скорости фотохимической реакции, объединяющее все законы фотохимии и дающее теоретическую интерпретацию коэффициенту пропорциональности в уравнении (3).
ОСНОВНЫЕ ТИПЫ ФОТОХИМИЧЕСКИХ ПРОЦЕССОВ.
Как показывает опыт, все фотохимические реакции в зависимости от величины квантового выхода можно подразделить на четыре группы :
1. Реакции, в которых квантовый выход g = 1 (например, образование перекиси водорода);
2. Реакции, в которых g < 1 (разложение аммиака, йодистого и бромистого метана, ацетона, уксусной кислоты);
3. Реакции, в которых g > 1 (образование хлористого сульфурила, бромистого водорода, озона, разложение бромистого водорода, двуокиси азота, азометана, хлорноватистой кислоты);
4. Реакции, в которых g >> 1 (реакция взаимодействия хлора с водородом).
Отклонение квантового выхода от единицы не означает отклонения от закона фотохимической эквивалентности. Фотохимический процесс слагается из первичного процесса, протекающего в результате поглощения светового кванта и, как правило, приводящего к диссоциации молекулы и образованию свободных атомов и радикалов, и из вторичных процессов, протекающих в результате вступления в реакцию образовавшихся атомов и радикалов. Первичные фотохимические процессы, являющиеся истинно фотохимическими, всегда подчиняются закону эквивалентности Штарка-Эйнштейна. Таким образом, отклонение квантового выхода от единицы означает появление вторичных процессов, которые идут уже без поглощения света.
Квантовый выход реакций, протекающих в растворах или в газах при очень малых давлениях, очень часто оказывается меньше 1. При реакциях в растворах это происходит вследствие дезактивации возбужденных молекул в результате передачи энергии молекулам растворителя или в результате рекомбинации возникших при фотодиссоциации атомов и молекул, причем рекомбинация облегчается молекулами растворителя, играющими роль третьих частиц. Такое уничтожение реакционноспособных частиц получило название эффекта ячейки (клетки) Франка-Рабиновича.
Если реакция идет в газах, находящихся под малым давлением, то возникшие активные молекулы могут дезактивироваться путем испускания света до того, как они столкнутся с реагирующими молекулами.
В ряде случаев квантовый выход оказывается больше 1; особенно часто он равен 2 или 3. Примерами таких процессов могут служить реакции разложения в газовой фазе следующих веществ (g = 2):
hn
2HJ ® H2 + J2
2HBr ® H2 + Br2
2NOCl ® 2NO + Cl2
Если Вам понравилась эта лекция, то понравится и эта – 22 Кризис Римской Империи.
Фотохимическое разложение бромистого и йодистого водорода представляет собой фотореакции, механизм которых наиболее известен.
В некоторых случаях квантовый выход может быть порядка десятков. Например, для реакции разложения перекиси водорода в воде квантовые выходы лежат в пределах от 7 до 500. Детальный механизм реакций, для которых g > 1, во многих случаях неизвестен.
Примером реакций, для которых g >> 1, является уже рассмотренная реакция соединения хлора с водородом на свету. Для этой реакции g » 105, т.е. одному поглощенному кванту соответствует около ста тысяч превратившихся молекул.
Таким образом, во всех фотохимических реакциях первичный фотохимический процесс подчиняется закону эквивалентности Штарка – Эйнштейна, а характер отклонения от этого закона позволяет разобраться во вторичных, не фотохимических процессах.
К первичным фотохимическим процессам близки так называемые сенсибилизированные реакции, в которых участвуют не те молекулы, которые непосредственно поглощают лучистую энергию, а соседние молекулы, которые сами по себе нечувствительны к излучению данной частоты и получают энергию от непосредственно поглощающих её молекул. Примером такого процесса является диссоциация молекулярного водорода в присутствии паров ртути. Известно большое число сенсибилизированных реакций. Кроме паров ртути сенсибилизаторами могут быть галогены, хлорофилл, ионы железа и др.
В фотохимических реакциях равновесие смещается под действием света. Заметное нарушение равновесия можно наблюдать только тогда, когда квантовый выход реакции близок к единице.
From Wikipedia, the free encyclopedia
In particle physics, the quantum yield (denoted Φ) of a radiation-induced process is the number of times a specific event occurs per photon absorbed by the system.[1]
Applications[edit]
Fluorescence spectroscopy[edit]
The fluorescence quantum yield is defined as the ratio of the number of photons emitted to the number of photons absorbed.[2]

Fluorescence quantum yield is measured on a scale from 0 to 1.0, but is often represented as a percentage. A quantum yield of 1.0 (100%) describes a process where each photon absorbed results in a photon emitted. Substances with the largest quantum yields, such as rhodamines, display the brightest emissions; however, compounds with quantum yields of 0.10 are still considered quite fluorescent.
Quantum yield is defined by the fraction of excited state fluorophores that decay through fluorescence:

where
- Φf is the fluorescence quantum yield,
- kf is the rate constant for radiative relaxation (fluorescence),
- knr is the rate constant for all non-radiative relaxation processes.
Non-radiative processes are excited state decay mechanisms other than photon emission, which include: Förster resonance energy transfer, internal conversion, external conversion, and intersystem crossing. Thus, the fluorescence quantum yield is affected if the rate of any non-radiative pathway changes. The quantum yield can be close to unity if the non-radiative decay rate is much smaller than the rate of radiative decay, that is kf > knr.[2]
Fluorescence quantum yields are measured by comparison to a standard of known quantum yield.[2] The quinine salt quinine sulfate in a sulfuric acid solution was regarded as the most common fluorescence standard,[3] however, a recent study revealed that the fluorescence quantum yield of this solution is strongly affected by the temperature, and should no longer be used as the standard solution. The quinine in 0.1M perchloric acid (Φ = 0.60) shows no temperature dependence up to 45 °C, therefore it can be considered as a reliable standard solution.[4]
| Compound | Solvent | 
|

|
|---|---|---|---|
| Quinine | 0.1 M HClO4 | 347.5 | 0.60 ± 0.02 |
| Fluorescein | 0.1 M NaOH | 496 | 0.95 ± 0.03 |
| Tryptophan | Water | 280 | 0.13 ± 0.01 |
| Rhodamine 6G | Ethanol | 488 | 0.94 |
Experimentally, relative fluorescence quantum yields can be determined by measuring fluorescence of a fluorophore of known quantum yield with the same experimental parameters (excitation wavelength, slit widths, photomultiplier voltage etc.) as the substance in question. The quantum yield is then calculated by:

where
- Φ is the quantum yield,
- Int is the area under the emission peak (on a wavelength scale),
- A is absorbance (also called “optical density”) at the excitation wavelength,
- n is the refractive index of the solvent.
The subscript R denotes the respective values of the reference substance.[5][6] The determination of fluorescence quantum yields in scattering media requires additional considerations and corrections.[7]
FRET efficiency[edit]
Förster resonance energy transfer efficiency (E) is the quantum yield of the energy-transfer transition, i.e. the probability of the energy-transfer event occurring per donor excitation event:

where
- kET is the rate of energy transfer,
- kf the radiative decay rate (fluorescence) of the donor,
- knr are non-radiative relaxation rates (e.g., internal conversion, intersystem crossing, external conversion etc.).[8][9]
Solvent and environmental effects[edit]
A fluorophore’s environment can impact quantum yield, usually resulting from changes in the rates of non-radiative decay.[2] Many fluorophores used to label macromolecules are sensitive to solvent polarity. The class of 8-anilinonaphthalene-1-sulfonic acid (ANS) probe molecules are essentially non-fluorescent when in aqueous solution, but become highly fluorescent in nonpolar solvents or when bound to proteins and membranes. The quantum yield of ANS is ~0.002 in aqueous buffer, but near 0.4 when bound to serum albumin.
Photochemical reactions[edit]
The quantum yield of a photochemical reaction describes the number of molecules undergoing a photochemical event per absorbed photon:[1]

In a chemical photodegradation process, when a molecule dissociates after absorbing a light quantum, the quantum yield is the number of destroyed molecules divided by the number of photons absorbed by the system. Since not all photons are absorbed productively, the typical quantum yield will be less than 1.

Quantum yields greater than 1 are possible for photo-induced or radiation-induced chain reactions, in which a single photon may trigger a long chain of transformations. One example is the reaction of hydrogen with chlorine, in which as many as 106 molecules of hydrogen chloride can be formed per quantum of blue light absorbed.[10]
Quantum yields of photochemical reactions can be highly dependent on the structure, proximity and concentration of the reactive chromophores, the type of solvent environment as well as the wavelength of the incident light. Such effects can be studied with wavelength-tunable lasers and the resulting quantum yield data can help predict conversion and selectivity of photochemical reactions.[11]
In optical spectroscopy, the quantum yield is the probability that a given quantum state is formed from the system initially prepared in some other quantum state. For example, a singlet to triplet transition quantum yield is the fraction of molecules that, after being photoexcited into a singlet state, cross over to the triplet state.
Photosynthesis[edit]
Quantum yield is used in modeling photosynthesis:[12]
See also[edit]
- Quantum dot
- Quantum efficiency
References[edit]
- ^ a b Braslavsky, S. E. (2007-01-01). “Glossary of terms used in photochemistry, 3rd edition (IUPAC Recommendations 2006)”. Pure and Applied Chemistry. 79 (3): 293–465. doi:10.1351/pac200779030293. ISSN 1365-3075. S2CID 96601716.
- ^ a b c d Lakowicz, Joseph R. Principles of Fluorescence Spectroscopy (Kluwer Academic / Plenum Publishers 1999) p.10. ISBN 978-0-387-31278-1
- ^ Brouwer, Albert M. (2011-08-31). “Standards for photoluminescence quantum yield measurements in solution (IUPAC Technical Report)”. Pure and Applied Chemistry. 83 (12): 2213–2228. doi:10.1351/PAC-REP-10-09-31. ISSN 1365-3075. S2CID 98138291.
- ^ Nawara, Krzysztof; Waluk, Jacek (2019-04-16). “Goodbye to Quinine in Sulfuric Acid Solutions as a Fluorescence Quantum Yield Standard”. Analytical Chemistry. 91 (8): 5389–5394. doi:10.1021/acs.analchem.9b00583. ISSN 0003-2700. PMID 30907575. S2CID 85501014.
- ^ Albert M. Brouwer, Standards for photoluminescence quantum yield measurements in solution (IUPAC Technical Report), Pure Appl. Chem., Vol. 83, No. 12, pp. 2213–2228, 2011. doi:10.1351/PAC-REP-10-09-31.
- ^ Levitus, Marcia (2020-04-22). “Tutorial: measurement of fluorescence spectra and determination of relative fluorescence quantum yields of transparent samples”. Methods and Applications in Fluorescence. 8 (3): 033001. Bibcode:2020MApFl…8c3001L. doi:10.1088/2050-6120/ab7e10. ISSN 2050-6120. PMID 32150732. S2CID 212653274.
- ^ Lagorio, María Gabriela (2020-10-06). “Determination of Fluorescence Quantum Yields in Scattering Media”. Methods and Applications in Fluorescence. 8 (4): 043001. Bibcode:2020MApFl…8d3001L. doi:10.1088/2050-6120/aba69c. ISSN 2050-6120. PMID 32674086. S2CID 220610164.
- ^ dos Remedios, Cristobal G.; Moens, Pierre D.J. (September 1995). “Fluorescence Resonance Energy Transfer Spectroscopy Is a Reliable “Ruler” for Measuring Structural Changes in Proteins”. Journal of Structural Biology. 115 (2): 175–185. doi:10.1006/jsbi.1995.1042. PMID 7577238.
- ^ “Fluorescence Resonance Energy Transfer”. Chemistry LibreTexts. 2013-10-02. Retrieved 2020-11-30.
- ^ Laidler K.J., Chemical Kinetics (3rd ed., Harper & Row 1987) p.289 ISBN 0-06-043862-2
- ^ Menzel, Jan P.; Noble, Benjamin B.; Blinco, James P.; Barner‐Kowollik, Christopher (2021). “Predicting wavelength-dependent photochemical reactivity and selectivity”. Nature Communications. 12 (1): 1691. Bibcode:2021NatCo..12.1691M. doi:10.1038/s41467-021-21797-x. PMC 7966369. PMID 33727558.
- ^ Skillman JB (2008). “Quantum yield variation across the three pathways of photosynthesis: not yet out of the dark”. J. Exp. Bot. 59 (7): 1647–61. doi:10.1093/jxb/ern029. PMID 18359752.
Первоначальные
законы фотохимии были установлены еще
в 19 веке. Гротгус (1817) и Дрейнер (1830)
независимо друг от друга сформулировали
закон , согласно которому “только те
лучи света могут химически действовать
на вещество, которые поглощаются этим
веществом”.Обратное утверждение,
что лучи, поглощаемые веществом, химически
активны, далеко не всегда справедливо
(энергия поглощенных квантов может быть
недостаточна для перевода вещества в
реакционноспособное состояние).
Действительно,
для протекания фотохимических реакций
необходимо, чтобы свет, входящий в
реакционный сосуд, поглощался реагирующим
соединением или сенсибилизатром.
Однако при количественных исследованиях
необходимо рассматривать все особенности
изучаемой реакции, нужно знать спектр
испускания источника света, спектры
поглощения фильтра, окошек сосуда,
растворителя, продуктов фотолиза.
В
1855г. Бунзен и Роско сформулировали закон
взаимозаместимости: “результат
фотохимического превращения зависит
от количества света, падающего на
светочувствительную систему, но не
зависит от того, как было подано это
количество света в виде большой
интенсивности за короткое время или в
виде малой интенсивности за длительное
время”. Если обозначить через I- интенсивность света и черезt– продолжительность освещения, можно
записать: результат фотохимических
реакций постоянен, еслиI1t1=I2t2= …=Iiti =const.
Однако
в ряде случаев (например, в фотографическом
процессе) наблюдаются существенные
отклонения от этого закона за счет
вторичных темновых реакций.
В
1904г. Вант-Гофф указал, что в законе
взаимозаместимости не учтено то, что
реакция может идти лишь под действием
света, поглощенного реакционной системой,
и сформулировал следующий закон:
«количество химически измененного
вещества (m) прямо
пропорционально количеству поглощенной
светом световой энергии (It)”.
Из закона следует, что скорость
фотохимических реакций (–dm/dt)
пропорциональна интенсивности
поглощенного светаI.
Использовав
закон Бугера-Ламберта-Беера, получим
выражение ( – dm/dt)
=KI0(1 – 10-εcl).
Это математическая запись закона
Вант-Гоффа.
Сформулированные
здесь законы являются следствием
основного закона фотохимии – закона
Эйнштейна (иногда закона Эйнштейна-Штарха).
В
1908-1912гг. Штарк и в 1912-1913гг. Эйнштейн
сформулировали закон квантовой
эквивалентности. Это основной закон
квантовой эквивалентности.Он известен
в нескольких формулировках. Наиболее
точная из них:каждый поглощенный
квант лучистой энергии вызывает активацию
одной молекулы или одного атома (другими
словами, число активированных молекул
равно числу поглощенных квантов).
Если
величина кванта достаточна для
активирования молекулы до реакционноспособного
состояния, то, очевидно, что число
реакционноспособных молекул равно
числу поглощенных квантов. В связи с
этим квантовый выход фотохимических
реакций всегда будет равен 1.
3.2. Квантовый выход фотохимической реакции
Под
квантовым выходом – фотохимических реакций понимается
отношение числа прореагировавших
молекул к числу поглощенных квантов:
=
число прореагировавших молекул/ число
поглощенных квантов = m/n.
Опыт
показывает, что далеко не во всех случаях
= 1.В большинстве случаев
1, но в ряде случаев
1.(Таблица 4).
Таблица
4.
Величины
квантового выхода некоторых фотохимических
реакций
|
Реакция |
|
, |
Светопоглощение |
Среда, |
|
1. |
1 |
172-173 |
O2 |
Газовая |
|
2. |
1 |
579 |
I2 |
Водный |
|
3. |
2 |
207-282 |
HI |
Газовая |
|
4. |
0,08-0,11 |
222-282 |
HI |
Водный |
|
5. |
3 |
170-190 |
O2 |
Газовая |
|
6. |
1000 |
400-436 |
Cl2 |
Газовая |
|
7. |
104-106 |
303-500 |
Cl2 |
Газовая |
|
8. |
0,01 |
202-210 |
CH3I |
Газовая |
Отклонение
квантовых выходов от единицы можно
объяснить следующим образом:
1.
Возбужденные светом молекула или атом
могут претерпевать не только фотохимические,
но и фотофизические превращения –
излучение кванта люминесценции или
тепловую деградацию энергии. Сумма
квантовых выходов всех этих первичных
процессов φi
=1, следовательно, квантовый выход
первичной фотохимической реакции может
быть или равен, или меньше1. Чем
больше доля физических превращений,
тем меньше квантовый выход.
2.
В большем случае продукты первичной
реакции претерпевают дальнейшие
изменения, т.е. за фотохимией, следует
темновая стадия реакции. Например,
образующиеся под действием света –
радикалы А→R•
+ R•, могут
соединиться обратно в молекулуА,
а могут начать цепную реакцию.
Наличие
темновых процессов приводит к двум
важным следствиям:
-
квантовый
выход реакции в целом может значительно
отличаться от 1 как в большую, так и в
меньшую сторону; -
квантовые
выходы одной и той же реакции могут
существенно различаться в зависимости
от того, при каких условиях и в какой
сфере проводится реакция. Так, например,
для реакции 2HI
H2 + I2в газовой сфере=2,а в водном растворе=0,8-0,11.
Другой пример фотолиз кристалловAgHalпри
облучении осадкаAgBrв водной среде актиничным светом(
= 400-420нм)
меньше 1 (0,2-0,3).
Однако если к раствору над осадком
добавитьNaNO2желатину
или другой бромакцептор (т.е. вещество,
поглощающее свободный бром), квантовый
выход фотолизаAgBr
Ag + Brподнимается почти до1(
1).
Бромакцептор связываетBr,
выделяемый при фотолизе, и тем самым
затрудняет (или делает невозможной)
обратную реакцию образованияAgBr:
Ag + Br
= AgBr. В отсутствие
бромакцептора обратная реакция протекает
в значительной степени, уничтожая
результаты действия света, в силу чего1.
Квантовый
выход меньше 1 может быть обусловлен не
только обратной реакцией, но и другими
причинами. Опыт показывает, что 1особенно часто наблюдается в случае
реакций, протекающих в растворах, а
также в газах при очень малых давлениях.
В случае реакции в растворах1обусловлен не только рекомбинацией,
возникших при облучении продуктов
реакции, но в значительной степени
дезактивацией возбуждающих молекул
молекулами-растворителями. В разреженных
газах (а также в кристаллических телах
при низких температурах), если реакция
идет с участием возбуждающих молекул,М + hv = М*,активные молекулы, прежде чем они придут
в контакт с другими молекулами, с которыми
они должны прореагировать, дезактивируются
путем самопроизвольного излучения
поглощенной энергии,М* = М + hv.
В
случае кристаллических тел при низких
температурах потеря энергии возбуждения
также идет путем люминесценции. В случае
более концентрированных газов и при
более высоких температурах кристаллических
тел явление люминесценции играет меньшую
роль, но возрастает вероятность
рекомбинации продуктов реакций, а также
потери энергии возбуждения активными
молекулами в результате столкновения
с аналогичными, но невозбужденными
молекулами.
Фотохимические
реакции протекают с квантовым выходом
больше 1 тогда, когда темновые процессы,
следующие за первичным, собственно
фотохимическим актом, имеют то же, (но
не обратное) направление, что и первичный
акт. Например, реакция 2HI
+hv
H2 + I2протекает в газовой среде с=2.Это обусловлено следующей последовательностью
реакций:
HI
+hv
H2 + I2– первичный фотохимический акт,=1;
Н•
+ HI
H2 + I•
– темновая реакция;
I•
+ I•
I2–
темновая реакция;
_________________________________________________________
2HI
+hv
H2 + I2– суммарная реакция,=2.
В
приведенных реакциях свободные атомы
HиIобладают
весьма высокой химической активностью,
т.к. являются, по существу, свободными
радикалами (отмечено точками). Особенно
высокий квантовый выход наблюдается
при протекании фотохимических цепных
реакций. Типичный пример таких реакций:
H2
+ Cl2
hν
2HCl,
=104– 106.
Реакция
протекает при облучении смеси Cl2иH2 синим
светом, что приводит к расщеплению
молекулCl2на атомы:Cl2
+ hv = 2Cl•(энергия квантов синей зоны недостаточна
для расщепления молекулы Н2, но
достаточна для диссоциации молекулCl2). Образующиеся
атомыCl• химически
весьма активны, поэтому при столкновении
с молекуламиH2протекает реакция:H2
+ Cl•
HCl + H•.
Образующиеся атомыH•
реагируют с молекулами Н•
+ Cl2
HCl + Cl•далее цикл повторяется. Таким образом,
поглощения первого кванта молекулойCl2ведет к расщеплению
ее на атомы и развитию двух цепей
темновых реакций с образованиемHCl:
Cl2
+ hv = Cl•
+ Cl•–
первичный акт,=1,
зарождение цепей;
+Н2/
HCl+Н•
+Н2/ HCl+Н•
– темновые процессы, развитие
цепей;
+Cl2/
HCl+ Cl•
+Cl2/
HCl+ Cl•
+Н2/
HCl+Н•
+Н2/ HCl+Н•– суммарное, =104-106
Из
приведенной схемы, казалось бы, следует,
что достаточно поглощения одного кванта
энергии для того, чтобы прореагировала
вся смесь. В действительности, помимо
развития цепей, имеет место их обрыв в
результате реакции:
Н•+Н•
Н2 + Q1
= 103,2ккал/моль; (1)
Cl•+
Cl•
Cl2 + Q2
= 57,1ккал/моль;.(2)
где
Q1, Q2
–теплоты соответствующих
реакцийQ1
= 103,2ккал/моль; Q2
= 57,1ккал/моль
Обрыв
цепей особенно легко происходит на
стенках сосуда, поэтому при прочих
равных условиях, квантовый выход этой
реакции тем больше, чем больше отношение
объема сосуда к суммарной поверхности
стенок. Обрыв цепей учащается также в
случае, если к реагируемой смеси H2
+ Cl2добавлен
какой-то газ, инертный по отношению к
этой реакции (например,N2).
Для сдвига реакций(1)и(2)в
сторону молекул необходимо поглощение
выделяемой энергии каким-то посторонним
телом, что и выполняют стенки сосуда
или молекулаN2
.
Знание
квантового выхода очень важно для
исследования механизма реакции и
определения ее скорости.
Если
заменить число поглощенных квантов и
света на интенсивность поглощения света
I,
а число прореагировавших молекул на
скорость изменения их концентрации –d[M]/dt,
то получим выражение: –d[M]/dt
=
Iα/Э
(Э– величина энергии моля квантов). Из
закона Бугера-Ламберта-Беера получим:d[M]/dt
=
I0/Э
(I – 10-ε c
l) выражение
для скорости фотохимических реакций,
объединяющее все законы фотохимии.
В
заключении необходимо отметить следующее
обстоятельство. Выше сформулирован
закон квантовой эквивалентности
Эйнштейна и указано, что число возбуждаемых
молекул равно числу поглощенных квантов,
на основании чего следует, что каждая
молекула, поглощающая лучистую энергию,
способна поглотить только один квант
энергии. Это соотношение действительно
соблюдается при обычных условиях
проведения фотохимических реакций
(квантовый выход первичного акта
фотохимической реакции равен единице).
Одноквантовость поглощения обусловлена
тем, что продолжительность пребывания
молекул в возбужденном состоянии очень
мала (как правило, 10-8-10-9с),
а обычно используемые интенсивности
облучения невысоки ( 1013– 1015квантов, поглощаемых 1 см3за 1 с).
поэтому концентрации возбужденных
молекул малы и поглощение ими еще одного
кванта маловероятно (аналогия с
вероятностью тройного соударения
молекул). Между тем, в настоящее время
в связи с развитием техники получения
потоков лучистой энергии весьма высокой
интенсивности (излучение лазеров,
техника импульсного фотолиза) стало
возможным проведение двухквантовых
процессов, т.е. таких фотохимических
реакций, в которых молекула переходит
в возбужденное состояние в результате
поглощения не одного, а двух квантов
энергии (так называемая двухквантовая
фотохимия).
Основная
литература (5
осн. [165-170])
Дополнительная
литература (4 [18-32])
Контрольные
вопросы:
-
Определение
основного закона фотохимии -
Понятие
«квантовый выход фотохимической
реакции» -
Причины
отклонения квантового выхода от единицы -
Влияние
условий и среды протекания фотохимической
реакции на квантовый выход. -
Причины
понижения квантового выхода реакции
(φ < 1). -
Виды
фотохимических цепных реакций. -
Значение
квантового выхода для определения
скорости фотохимической реакции.
Лекция
№4.
Поглощение лучистой энергии атомами и
молекулами
При
поглощении атомами и молекулами
ультрафиолетового (УФ) и видимого света
происходят переходы электронов на более
высокие энергетические уровни, в
результате чего наблюдается типичный
электронный спектр поглощения.
Поглощение
света происходит лишь в случае, если
разность между двумя энергетическими
уровнями в точности соответствует
энергии кванта:
Δ
Е = h ·
ν = Е2
– Е1,
где
Е2 и Е1 –
энергия молекулы в конечном и начальном
состоянии соответственно.
Если
энергия начального состояния меньше
энергии конечного Е1<
Е2 , то молекула или атом
поглощают квант лучистой энергии. При
обратном соотношенииЕ1 >
Е2 квант излучается.
Таким образом, возникают спектры
поглощения или испускания. Явления,
сюда относящиеся, известны также под
названием поглощения, резонанса,
флуоресценции, фосфоресценции одноатомных
паров и газов и заключается в том, что
данный газ, помещенный на пути пучка
монохроматического света, частоту
которого можно произвольно и непрерывно
менять, при достижении ею некоторых
определенных и характерных для элемента,
составляющего газ, значений начинает
испускать свет той же и других частот,
а возбуждающий проходящий свет при этом
ослабляется.
Молекулу
вещества (например, красителя) в
нормальном, или основном, состоянии с
энергией Е0, обозначим через А, а
молекулу, переведенную поглощением
фотонаhνна более высокий
уровень энергии Е* назовем возбужденной
и обозначим через А*.
В
отличие от основного возбужденное
состояние молекулы неустойчиво и имеет
весьма краткую продолжительность
существования. По истечении в среднем
10-9с (и короче) большинство
одновременно возбужденных молекул А*
возвращается в исходное основное
состояние и вновь может поглотить квант
света. Многие красители, растворенные
в жидких или твердых средах, поглощая
свет, дают интенсивное свечение, не
испытывая при этом никаких химических
изменений. Наличие такого явленияфлуоресценцииозначает, что энергия,
освобождаемая при возвращении возбужденной
молекулы в основное состояние, излучается
в виде фотонаhνf
. Рассматриваемые элементарные
процессы поглощения и испускания
световой энергии могут быть записаны
в виде следующих физических реакций:
поглощение
hνa
+ А → А*
испускание
А* → А + hνf
Спектр
испускания красителей представляет
собой широкую полосу, занимающую в
спектре, как и полоса поглощения,
протяжение порядка 100 нм, но всегда
сдвинутую по отношению к последней в
сторону длинных волн. Такова закономерность
явления флуоресценции, которая обнаружена
и сформулирована под названием правила
Стокса, согласно которому частота
излучаемого света νf
должна быть всегда меньше частотыνa
света поглощенного:
hνf<
hνa
Это
означает, что в виде фотона всегда
испускается только часть поглощенного
кванта. Остаток hνa
-hνf
задерживается в молекуле в виде
избыточной термической энергии, быстро
отдаваемой окружению.
Функциональные
группы, поглощающие электромагнитное
излучение в УФ и видимой областях спектра
называются хромофорами. Типичными
хромофорами являются:
С=С
(λ макс= 175;185 нм),![]() (λмакс= 160;185; 280 нм)
(λмакс= 160;185; 280 нм)
Функциональные
группы (например, ОН, СООН, NH2,OR,NO2),
которые вступают в сопряжение с
хромоформами за счет своих неподеленных
пар электронов с образованием новых
хромофоров, называютауксохромами.
Ультрафиолетовая
и видимая спектроскопия дает информацию
о наличии двойных или тройных
углерод-углеродных связей, ароматических
колец, азот-, кислород- и серосодержащих
или других группировок, для которых
характерны σ → σ*, π→π* и т.д. Обычно
УФ и видимые спектры снимают в очень
разбавленных растворах (10-4-10-6моль/л), естественно в исследуемом
диапазоне длин волн растворитель не
должен иметь полосу поглощения.
При
поглощении инфракрасного излучения
(ИК-излучения) молекулы вещества переходят
в колебательно-возбужденное состояние.
Частоты колебаний атомов в молекуле,
а, следовательно, и частоты поглощенного
излучения, определяются природой атомов,
их расположением в молекуле и характером
связей. Так как молекулы разных веществ
различаются по этим показателям, то и
инфракрасные спектры сугубо характерны
для каждого вещества.
Важной
особенностью ИК-спектров является то,
что группы атомов имеют характерное
расположение полос поглощения в спектре,
то есть определенные характеристические
полосы. Поэтому по ИК-спектрам можно не
только идентифицировать вещество, но
и получить сведения о составе и строении
неизвестного вещества.
В
настоящее время определены характеристические
частоты ИК-спектров многих веществ и
групп атомов. Оказалось, что
характеристические частоты групп атомов
не имеют строго фиксированной величины,
а лежат в довольно узких диапазонах,
внутри которых обнаруживается поглощение
этих групп в большом числе химических
соединений.
Частоты
излучений в ИК-спектроскопии принято
выражать в волновых числах ω = 1/λ. Единицей
измерения волнового числа является
см-1. Волновое число пропорционально
частоте
ω
= ν/с, где ν – частота в с-1, с –
скорость света в см/с. Полосы поглощения,
наиболее характерные для молекулярных
структур, лежат в интервале 5000-650 см-1,
что соответствует длинам волн 2-15 мкм.
ИК-спектры
можно использовать для качественного
и количественного анализа веществ и
их превращений, в том числе и для изучения
фотохимических реакций. Строят
спектральные кривые в координатах α %
= f(ω) и τ % =f(ω). На кривых наблюдается система
максимумов (пиков) поглощения, имеющих
расположение, характерное для данного
вещества. Пики поглощения соответствуют
максимумам на кривых α % =f(ω) и минимумам на кривых τ % =f(ω). Волновые числа, соответствующие
пикам поглощения называются
характеристическими частотами.
Характеристические частоты используются
для качественного анализа вещества, а
высота пиков – для количественного.
При
качественном анализе, например, при
идентификации неизвестного вещества,
проводят сравнение его характеристических
частот со справочными данными по спектрам
поглощения различных функциональных
групп. Если в спектре поглощения
исследуемого неизвестного вещества
есть максимумы в области характеристических
частот данной функциональной группы,
можно сделать заключение о наличии
данной группы в молекуле. Необходимо
учитывать, что наличие растворителя,
образование водородных связей,
кристаллизация могут привести к сдвигу
максимума в спектре. Это затрудняет
анализ неизвестного вещества и является
достоинством при анализе вещества
известного, так как позволяет получить
о нем дополнительные сведения.
При
фотохимических превращениях пики
поглощения, характерные для реагента,
начинают уменьшаться и при полном
расходовании вещества исчезают. Пики,
соответствующие продуктам реакции,
наоборот, появляются и растут. Изменение
спектров поглощения вещества позволяет
судить об изменениях, происходящих в
нем в процессе фотохимической реакции.
В
таблице 5 приведены характеристические
частоты некоторых функциональных групп
фоторезистов, применяемых в полиграфической
промышленности и электронной технике.
Таблица
5
Характеристические
частоты химических групп
|
Функциональная |
Характеристическая |
|
Группа -О-СО-СН=СНС6Н5 |
1635 |
|
Группа |
2100-2160 |
|
Группа |
1600-1620 |
|
Группа |
2124 |
Высота
пиков поглощения в спектре вещества
(на спектральной кривой) определяется
его количеством в системе. В случае
соблюдения закона Бугера-Ламберта-Беера
по высоте пика можно подсчитать
концентрацию вещества, используя
формулу:lgτλ= – ελcl, где с –
концентрация вещества в таблетке или
пленке, ελ – молярный коэффициент
поглощения,l– толщина
образца.
Если
исследуется фотохимическая реакция,
происходящая в пленке, то концентрацию
реагента или продукта в пленке в данный
момент времени можно подсчитать по
формуле:
Сх= С0lgτх
/τ0, гдеτ0
– коэффициент пропускания пленкой
излучения с характеристической частотой
для известной концентрации вещества,
такой концентрацией может быть начальная
С0;
τх
– коэффициент пропускания для текущей
концентрации (через времяtхот начала реакции).
Так
как по ИК-спектрам можно определить
количество образовавшегося при реакции
продукта, то их можно использовать для
определения квантового выхода. По
спектрам можно также рассчитать константу
скорости фотолиза. Так как фотолиз
о-нафтохинондиазида – реакция первого
порядка, для него константу скорости
фотолиза Кфможно определить по
формуле:
Кф=dlnC/dT. Перейдя по закону
Бугера-Ламберта-Беера к оптической
плотности, а затем к коэффициенту
пропускания получаем Кф= ΔlnD/Δt, где Δt– интервал времени
фотолиза, в течение которого высота
пика изменилась с τ0τх. Переход
от τ кDосуществляется
по формулеD= -lgτ
Основная
литература (2 [21-33])
Дополнительная
(5 [24-75])
Контрольные
вопросы:
-
Условия,
необходимые для поглощения (испускания)
света веществом -
Правило
Стокса -
Понятие
флуоресценции -
Определение
понятий «хромофоры», «ауксохромы» -
Характеристические
частоты в ИК-спектрах -
ИК-спектры
для идентификации вещества
Лекция
№5.Атомные и молекулярные спектры
Как
было указано в предыдущих лекциях,
начальный акт любой фотохимической
реакции заключается в поглощении кванта
лучистой энергии, в результате чего
запас энергии в поглощаемой системе
(атоме, молекуле, кристалле и др.)
возрастает: Е0+hν= Е*.
Что
же происходит в атомах и молекулах в
результате поглощения лучистой энергии?
Любой
квантовый переход вещества, заключается
в поглощении ли испускании кванта
лучистой энергии, определяется основным
уравнением частот Бера: hv
= hc/
= E
– E.
Е0
+ hv = E*
E*
+
hv
Е0
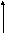



Рисунок
5.1. Уровни энергии Е0
и Е до и
после поглощения системой кванта
лучистой энергии
Если
энергия начального состояния меньше
энергии конечного: EE,то молекула или атом поглощают квант
лучистой энергии. При обратном соотношении:EEквант излучается. Таким образом, возникают
спектры поглощения или испускания.
Каждому квантовому переходу соответствует
одна спектральная линия (светящаяся
при испускании и темная при поглощении
квантов).Атомные спектрыимеют
линейчатый и относительно простой
характер, но поскольку каждый электронный
энергетический уровень, характеризуемый
квантовым числом –n,
может быть расщеплен на подуровни (s,
p, d,
f), характеризуемыми
побочными квантовыми числами –l,
спектральные линии могут быть также
расщеплены на более узкие, тесно
прилегающие линии.Молекулярные
спектрызначительно сложнее; они
наблюдаются в виде более или менее
широких полос, распадающихся при
достаточной разрешающей силе спектрального
прибора на совокупность тесно расположенных
линий. Сложность молекулярных спектров
по сравнению с атомами обусловлена тем,
что молекула, помимо электронных уровней,
обладает также дискретными колебательными
или вращательными уровнями, отражающими
колебательные движения ядер атомов,
составляющих молекулу, и энергию вращения
молекулы, как в целом, так и в отдельных
ее группировках (в случае сложных
органических молекул).
Энергетический
интервал между электронными уровнями
(как в атомах, так и в молекулах) составляет
примерно от 250 до 38 ккал/моль, что
соответствует длинам волн поглощаемого
или излучаемого света ()-
приблизительно от 100 до 750 нм. Колебательные
уровни лежат в интервале от нескольких
(1-10) ккал/моль до десятых долей ккал/моль,
что соответствуетпорядка 5-30мкм (ИК-область) разница между
вращательными уровнями еще меньше от
долей кал/моль до нескольких десятков
кал/моль, что соответствует
в далекой ИК-области (порядка 1000-10
000мкм). Очевидно, собственно колебательные
или собственно вращательные переходы
могут быть вызваны поглощением
ИК-излучения и обнаруживаются
соответственно в виде ИК-спектров, в то
время как к электронным переходам
относятся видимая и УФ-области спектра.
Поэтому, казалось бы, молекулярные
спектры в видимой и УФ-областях должны
быть хотя и значительно более сложными,
чем атомарные, но все же линейчатыми
(соответственно расстояниям между
электронными уровнями энергии). В
действительности они полосатые, т.е. в
определенных интервалах – сплошные
(отдельные линии лежат так близко друг
к другу, что спектральный прибор не в
состоянии их разрешить, т.е. передать
раздельно). Это объясняется следующим
образом.
Представим
себе простейший случай: 2-х атомную
молекулу. На рисунке 5.2 изображены (без
соблюдения точного масштаба) электронные,
колебательные и вращательные уровни
(обозначаемые э, к, в) этой молекулы.
Когда молекула находится в “замороженном
“ состоянии, т.е. совершенно не обладает
энергией возбуждения, в ней отсутствуют
колебательные и вращательные движения,
и уровень ее энергии определяется
некоторым исходным “нулевым” уровнем
электронной энергии (линии 0-0). Однако
достаточно молекуле поглотить некоторое
количество тепловой энергии, чтобы
уровень ее энергии поднялся на некоторую
большую или меньшую высоту, определяемую
соответственно колебательными и
вращательными уровнями. Когда мы имеем
достаточно большую совокупность молекул,
эта совокупность характеризуется
некоторым определенным распределением
энергий между молекулами, характерным
для данных условий.
В
результате поглощения лучистой энергии
будут иметь место электронные переходы,
характеризуемые величинами E
+hv =E,причем каждому из них будет соответствовать
определенная спектральная линия. Но
так как переходы будут совершаться не
только с данного основного (например,
нулевого) уровня, но с целого ряда
близлежащих колебательных и вращательных
уровней и будут они завершаться не
только на том или ином основном вышележащем
электронном уровне (например, на уровне
1), но также на многих других близлежащих
колебательных уровнях. Спектральная
линия существенно расширится и превратится
в спектральную полосу. Понятно, что чем
больше и сложнее молекула, тем больше
вероятность расширения спектральной
полосы и тем больше возможность слияния
отдельных полос в сплошной спектр,
характерный для данной системы (поэтому
конденсированные системы имеют сплошные
спектры поглощения).
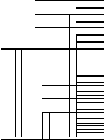
1
Э 1Э
3
к в
2 к в
1к
в
0
0
Рисунок
5.2 . Схема электронных, колебательных
и вращательных уровней молекулы.
Представим
теперь, что лучистая энергия воздействует
на какие-то свободные атомы (т.е. атомы,
не связанные или очень слабо связанные
с другими атомами). Например, пары Hgосвещаются УФ-излучением. АтомыHgпоглощают лучи тех длин волн,
которые удовлетворяют условию:
= hc/ E
– E.
В
случае атома речь может идти о возбуждении
только валентных уровней (вращательные
и колебательные уровни у свободных
атомов отсутствуют), т.е. в уравнении:
=
hc/ E
– E,
величиныE
– Eпредставляют собою разности энергии
разрешенных электронных уровней. В
результате поглощения соответствующего
кванта электрон может перейти на более
высокий энергетический уровень – атом
будет возбужденА + hv
= А*, или, если энергия кванта будет
достаточно велика, электрон может быть
вырван из атома, т.е. произойдетионизацияатома:
А
+ hv = А+е–.
(В атомном спектре это соответствует
коротко-волновой границе линий данной
спектральной серии). Время перехода
электрона с одного уровня на другой
составляет 10-15-10-16с,
продолжительность существования
возбужденного состояния атома 10-7-10-9
с. Таким образом, хотя время пребывания
атома в возбужденном состоянии ~ на 8
порядков больше времени возбуждения,
оно все же ничтожно мало; возбужденный
атом очень быстро теряет энергию
возбуждения и это может произойти двумя
разными путями. Если возбужденный атом
за указанное время не встретится с
каким-то другим атомом и не передаст
ему свою избыточную энергию, электрон
с более высокого энергетического уровня
перейдет на более низкий (рис.5.3) основной
или промежуточный уровень. Этот переход
(падение электрона на более низкий
уровень) будет сопровождаться выпадением
кванта лучистой энергии: А* А+hv,
т.е. будет иметь место явлениелюминесценции
(в зависимости от длительности свечения
люминесценция делится на флюоресценцию
(10-8– 10-9с) и фосфоресценцию
(секунды, минуты, а иногда и часы)). Однако,
если возбуждающий атомА*прежде,
чем он потеряет избыточную энергию,
встретится с каким-то другим атомом или
молекулой (М) и передаст им свою
избыточную энергию, то атом или молекула,
которым была передана энергия возбуждения
(М*),могут претерпевать физические
или химические превращения:
А
432 1
*+МА+М*
М*– химические или физические превращения.
Подобные фотохимические реакции, в
которых одно вещество, поглощая лучистую
энергию, переходит в возбуждающее
состояние, но саму энергию возбуждения
не использует, а передает ее другому
веществу, которое и претерпевает
химическое изменение, называют
сенсибилизированными реакциями. Примером
такой реакции может служить
сенсибилизированный фотолиз водорода.
Кванты УФ-излучения с300нм
несут достаточную энергию, для размещения
молекул Н2на атомы, но облучение
газообразногоН2 при=254нм не вызовет
разложение молекулН2,
т.к. они это излучение не поглощают.
Р исунок
исунок
5.3. Схема энергетических переходов при
поглощении и испускании (люминесценция)
кванта лучистой энергии
Однако,
если к Н2 добавить
немного паровН2,молекулы Н2под действием излучения
с=254нм расщепляются
на атомы. Процесс идет по следующей
схеме:
Hg+hv
Hg*; Hg*+
Н2
Hg+ Н2*;
Н2*
2Н
Поглощение
лучистой энергии молекулами подчиняется
тем же закономерностям, что и поглощение
атомами. Поскольку молекула значительно
более сложная система, определяющаяся
рядом дополнительных условий, разрешающих
или запрещающих соответствующие
электронные переходы.
Для
описания электронной структуры молекул
в квантовой механике используют метод
линейной комбинации атомных орбит (ЛКАО
МО). Молекулярная орбиталь, как и атомная,
характеризуется набором четырех
квантовых чисел n,
l, m,
s; гдеn
– главное квантовое число
– определяет уровень энергии, на котором
находится электрон (может принимать
значения любого положительного целого
числа),l
– орбитальное квантовое число
определяет форму электронного облака
(может принимать значения положительных
целых чисел от 0 до n-1
включительно), m
– магнитное квантовое число– определяет положение молекулярной
орбитали относительно оси молекулы
(определяемой проекцией вектора
орбитального момента на межъядерную
ось в магнитном поле) т.е. определяет
ориентацию электронного облака в
пространстве. Может принимать значения
целых чисел от –lдо +l,
включая ноль.s –
спиновое квантовое число, определяющее
собственный момент количества движения
электрона, может принимать значения
+1/2 и –1/2.
Для
определения состояния электрона в
многоэлектронном атоме важное значение
имеет сформулированное В. Паули положение
(принцип Паули), согласно которому в
атоме не может быть двух электронов, у
которых все 4 квантовых числа были бы
одинаковыми. Из этого следует, что
каждая атомная орбиталь, характеризующаяся
определенными значениями квантовых
чисел, может быть занята не более чем
двумя электронами, спины которых имеют
противоположные знаки.
Молекулярная
орбиталь, как и атомная, не может быть
занята более чем 2-мя электронами, причем
эти 2 электрона должны обладать
противоположными спинами.
При
образовании простых (двухатомных)
молекул из атомных s-орбиталей
образуются σ-орбиталь, из р-орбиталей
образуется π-орбиталь, изd-орбиталей
δ-орбиталь. При комбинировании атомных
орбиталей могут образовываться двоякого
рода молекулярные орбитали: связывающие
и разрыхляющие. В связывающей орбитали
электронная плотность между ядрами
атомов высока, что вызывает взаимное
притяжение двух ядер, таким путем
образуется ковалентная химическая
связь между атомами. В разрыхляющей
орбитали электронная плотность между
ядрами понижена или равна нулю, вследствие
чего положительно заряженные ядра
взаимно отталкиваются. Разрыхляющая
орбиталь имеет более высокую энергию,
чем связывающая орбиталь, поэтому
перевод электрона со связывающей
орбитали на разрыхляющую орбиталь
требует затраты энергии (другими словами,
в этом случае требуется, чтобы молекула
получила определенную порцию энергии
извне, например, в виде кванта лучистой
энергии). Перевод электрона со связывающей
орбитали на разрыхляющую орбиталь
приводит к разрыву химической связи.
Изучение
атомных спектров показывает, что их
линии часто имеют тонкую структуру,
т.е. расщеплена на 2,3 или более близлежащих
линий.
Число
линий в тонкой структуре называют
мультиплетностью. Мультиплетность
отражает число возможных состояний
валентных электронов и может быть
рассчитана по формуле:
М=2S+1,
гдеS– это суммарный
спин атома, равен сумме спиновых квантовых
чисел валентных электронов. Если в
валентной электронной оболочке атома
или молекулы все электроны спарены, то
сумма их спиновых квантовых чисел =0. В
этом случаеМ=2*0+1=1,такое состояние
атома или молекулы называютсинглетным.
Если
у атома или молекулы имеется 1 неспаренный
электрон, то М=2*1/2+1=2. Такое состояние
называютдублетным.
При
наличии 2-х неспаренных электронов,
спины которых параллельны, мультиплетность
М=2(1/2+1/2)+1=3. Такое состояние атома
или молекулы называюттриплетным.
Если в синглетном состоянии атомы или
молекулы химически малоактивны, то в
дублетном и триплетном состояниях они
весьма реакционноспособны.
Синглетное
состояние обозначают буквой S,а триплетное – с индексом, указывающим
на энергетический уровень, на котором
находятся электроны. Например,S0
иT0–
соответственно синглетное и триплетное
состояние основного низшего энергетического
уровня,S1иT1 –
синглетное и триплетное состояние
первого возбужденного уровня и т.п.
Основная
литература (5 осн. [165-170])
Контрольные
вопросы:
1.
Поглощение и излучение лучистой энергии
атомами и молекулами.
-
2.
Электронный, колебательный и вращательный
уровни энергии. -
3.
Виды люминесценции -
4.
Квантовые числа, характеризующие
состояние атомов -
5.
Принцип Паули -
6.
Понятие мультиплетности -
7.
Синглетное, дублетное и триплетное
состояние. -
8.
Использование метода молекулярных
орбиталей для описания -
электронной
структуры молекул
Квантовый выход (Φ) из излучения , индуцированного процесса является количеством раз , когда событие происходит специфическое за фотон , поглощенного системой.
Приложения
Флуоресцентная спектроскопия
Квантовый выход флуоресценции определяется как отношение числа фотонов , испускаемых числу фотонов , поглощенных.

Квантовый выход флуоресценции измеряется по шкале от 0 до 1,0, но часто выражается в процентах. Квантовый выход 1,0 (100%) описывает процесс, в котором каждый поглощенный фотон приводит к испусканию фотона. Вещества с наибольшим квантовым выходом, такие как родамины , демонстрируют самые яркие излучения; однако соединения с квантовым выходом 0,10 по-прежнему считаются достаточно флуоресцентными.
Квантовый выход определяется долей флуорофоров в возбужденном состоянии , распадающихся за счет флуоресценции:

где – квантовый выход флуоресценции, – константа скорости радиационной релаксации (флуоресценции), – константа скорости всех безызлучательных релаксационных процессов. Безызлучательные процессы представляют собой механизмы распада возбужденного состояния, отличные от излучения фотонов, которые включают: резонансный перенос энергии Ферстера , внутреннее преобразование , внешнее преобразование и межсистемное пересечение . Таким образом, на квантовый выход флуоресценции влияет изменение скорости любого безызлучательного пути. Квантовый выход может быть близок к единице, если скорость безызлучательного распада намного меньше, чем скорость радиационного распада, то есть .




Квантовые выходы флуоресценции измеряются путем сравнения со стандартом известного квантового выхода. Хинин соль хинин сульфат в серной кислоте раствора рассматривается в качестве наиболее общего стандарта флуоресценции, однако, недавнее исследование показало , что квантовый выход флуоресценции данного раствора не сильно зависят от температуры, и больше не должен быть использован в качестве стандартного раствора . Хинин в 0,1 М хлорной кислоте ( = 0,60) не имеет температурной зависимости до 45 ° C, поэтому его можно рассматривать как надежный стандартный раствор.
| Сложный | Растворитель |
|
|
|---|---|---|---|
| Хинин | 0,1 млн 
|
347,5 | 0,60 ± 0,02 |
| Флуоресцеин | 0,1 млн
|
496 | 0,95 ± 0,03 |
| Триптофан | Воды | 280 | 0,13 ± 0,01 |
| Родамин 6G | Спирт этиловый | 488 | 0,94 |
Экспериментально относительный квантовый выход флуоресценции может быть определен путем измерения флуоресценции флуорофора с известным квантовым выходом с теми же экспериментальными параметрами ( длина волны возбуждения , ширина щели, напряжение фотоумножителя и т. Д.) , Что и рассматриваемое вещество. Затем квантовый выход рассчитывается по формуле:

где квантовый выход, Int представляет собой площадь под пиком излучения (по шкале длин волн), является поглощение (также называемым «оптическая плотностью») при длине волны возбуждения, а п является показателем преломления от растворителя . Нижний индекс R обозначает соответствующие значения эталонного вещества. Определение квантовых выходов флуоресценции в рассеивающих средах требует дополнительных рассмотрения и исправлений.
FRET эффективность
Фёрстеровский резонансный перенос энергии ( ) – это квантовый выход перехода с переносом энергии, т. Е. Вероятность возникновения события передачи энергии в расчете на одно событие возбуждения донора:


где – скорость передачи энергии, скорость радиационного распада (флуоресценции) донора и – скорости безызлучательной релаксации (например, внутреннее преобразование, межсистемное пересечение, внешнее преобразование и т. д.).

Растворители и воздействие на окружающую среду
Окружение флуорофора может влиять на квантовый выход, обычно в результате изменения скорости безызлучательного распада. Многие флуорофоры, используемые для мечения макромолекул, чувствительны к полярности растворителя. Класс молекул зондов 8-анилинонафталин-1-сульфоновой кислоты (ANS) практически не флуоресцирует в водном растворе, но становится сильно флуоресцентным в неполярных растворителях или при связывании с белками и мембранами. Квантовый выход ANS составляет ~ 0,002 в водном буфере, но около 0,4 при связывании с сывороточным альбумином .
Фотохимические реакции
Квантовый выход фотохимической реакции описывает количество молекул, подвергающихся фотохимическому событию, на поглощенный фотон:
В процессе химической фотодеградации , когда молекула диссоциирует после поглощения кванта света , квантовый выход представляет собой количество разрушенных молекул, деленное на количество фотонов, поглощенных системой. Поскольку не все фотоны продуктивно поглощаются, типичный квантовый выход будет меньше 1.
Квантовые выходы больше 1 возможны для фотоиндуцированных или радиационно-индуцированных цепных реакций , в которых одиночный фотон может запускать длинную цепочку превращений . Одним из примеров является реакция водорода с хлором , в которой на квант поглощенного синего света может образоваться до 10 6 молекул хлористого водорода .
В оптической спектроскопии квантовый выход – это вероятность того, что данное квантовое состояние образуется из системы, изначально приготовленной в каком-то другом квантовом состоянии. Например, квантовый выход синглетного перехода в триплетный – это доля молекул, которые после фотовозбуждения в синглетное состояние переходят в триплетное состояние.
Фотосинтез
Квантовый выход используется при моделировании фотосинтеза :
Смотрите также
- Квантовая точка
- Квантовая эффективность
