Гравиметрическое определение вещества в навеске соли
Вычислние навеси исследуемого образца сульфата меди
Задача 58.
Вычислить навеску исследуемого образца, необходимого для получения определенного количества весовой формы. Исследуемый образец: CuSO4; определяемое вещество CuSO4, приблизительное содержание 30 %; весовая форма BaSO4, масса 0,2г.
Решение:
Расчёт навески вещества в пробе проводим по формуле:

где а – масса весовой формы определяемого вещества, г;
f- аналитический множитель (фактор пересчета);
a – масса навески, г;
Р – приблизительное процентное содержание вещества в образце.
Mr(CuSO4) = 159,6;
Mr(BaSO4) = 233,39;
f = Mr(CuSO4)/Mr(BaSO4) = 159,6/233.39 = 0,6838.
Подставим все числовые значения в расчётную формулу:

Ответ: Для гравиметрического определения CuSO4 необходимо взвесить навеску CuSO4 массой близкой к 0,45г.
Вычислние навеси исследуемого образца хлорида марганца
Задача 59.
Вычислить навеску исследуемого образца, необходимого для получения определенного количества весовой формы. Исследуемый образец: МnСl2; определяемое вещество МnСl2, приблизительное содержание 32%; весовая форма AgCl, масса 0,3г.
Решение:
Расчёт навески вещества в пробе проводим по формуле:
где а – масса весовой формы определяемого вещества, г;
f- аналитический множитель (фактор пересчета);
a – масса навески, г;
Р – приблизительное процентное содержание вещества в образце.
Мr(MnCl2) = 125,844;
Mr(AgCl) = 143,321;
f = Мr(MnCl2)/2Mr(AgCl) = 125,844/(2 . 143,321) = 0,4390.
Подставим все числовые значения в расчётную формулу:

Ответ: Для гравиметрического определения МnСl2необходимо взвесить навеску МnСl2 массой близкой к 0,41г.
37
|
Vконц |
c разб |
Vразб |
. |
(2.13 в) |
|
|
сконц |
|||||
Пример 2.4. Исследуемый образец уксусной кислоты имеет плотность 1,015 г/см3, которой соответствует концентрация 1,98 моль/дм3. Для титрования предполагается использовать 0,1М раствора NaOH. Очевидно, что концентрация исследуемого образца значительно больше, чем у титранта. Рассчитывают объем исследуемого концентрированного раствора, необходимый для приготовления, к примеру, 200,0 см3 примерно 0,1М СН3СООН (разбавленный раствор)
|
V |
0,1 |
200,0 |
10,0 см3 . |
|
|
конц |
||||
|
1,98 |
||||
Когда объем концентрированного раствора больше или равен 5 см3, его можно измерить с помощью точной пипетки (в данном случае на 10,00 см3); если объем небольшой, то его измеряют мерным цилиндром (или пробиркой с делениями) переносят во взвешенный бюкс, определяют точную массу и количественно переносят навеску в мерную колбу.
При достаточно большой эквивалентной массе исследуемого вещества определение проводят методом отдельных навесок. Массу навески рассчитывают по формуле 2.8 а.
Пример 2.5. Рассчитать массу навески соли Мора (NH4)2Fe(SO4)2 · 6H2O, чтобы на ее титрование было затрачено 20,0 см3 0,05н. раствора KMnO4. Титрование соли железа раствором перманганата калия описывается уравнением реакции:
5Fe2+ + MnO4– + 8H+ = 5Fe3+ + Mn2+ + 4H2O.
Фактор эквивалентности железа равен 1, молярная масса соли Мора равна
392,13 г/моль.
|
0,05 |
392,13 |
20,0 |
||
|
m( соли Мора) |
0,392 г. |
|||
|
1000 |
||||
Формулы расчета массы навески твердого вещества представлены в таблице 2.2.
Таблица 2.2
Формулы расчета массы навески и результатов титриметрического анализа при прямом титровании
|
Способ |
|||
|
титро- |
Способ отдельных навесок |
||
|
вания |
|||
|
Концен- |
|||
|
трация |
масса навески, г (mобщ) |
массовое содержание компонента А в |
|
|
титран- |
образце, г (m) |
||
|
та |
|||
|
Т(В/А) |
mобщ = T(B/A) V(B) |
m = T(B/A) V(B) |
|
38
|
с[(1/z)B] |
mобщ |
с[(1/z)B] M(A) f экв (A) V(B) |
m |
c[(1/z)B] M(A) f экв (A) V(B) |
||||||||||||||||
|
1000 |
||||||||||||||||||||
|
1000 |
||||||||||||||||||||
|
T(B) |
mобщ |
T(B) M(A) fэкв(A)V(B) |
m |
T(B) M(A) f экв (A) V(B) |
||||||||||||||||
|
M(B) fэкв( В) |
M(B) f экв (B) |
|||||||||||||||||||
|
Способ |
||||||||||||||||||||
|
титро- |
Способ пипетирования |
|||||||||||||||||||
|
вания |
||||||||||||||||||||
|
Концен- |
||||||||||||||||||||
|
трация |
масса навески, г (mобщ) |
массовое содержание компонента А в |
||||||||||||||||||
|
титран- |
образце, г (m) |
|||||||||||||||||||
|
та |
||||||||||||||||||||
|
Т(В/А) |
mобщ = T(B/A) Vобщ |
m = T(B/A) V(B) |
Vобщ |
|||||||||||||||||
|
Va |
||||||||||||||||||||
|
с[(1/z)B] |
mобщ |
с[(1/z)B] M(A) f экв (A) Vобщ |
m |
c[(1/z)B] M(A) fэкв (A) V(B) Vобщ |
||||||||||||||||
|
1000 |
1000 |
V |
||||||||||||||||||
|
a |
||||||||||||||||||||
|
T(B) |
mобщ |
T(B) M(A) fэкв (A) Vобщ |
m |
T(B) M(A) fэкв (A) V(B) Vобщ |
||||||||||||||||
|
M(B) fэкв (B) |
Va |
|||||||||||||||||||
|
M(B) fэкв (B) |
В – титрант, А – определяемое вещество, Vобщ – объем мерной колбы, Vа – объем аликвоты, mобщ – масса навески, m – массовое содержание компонента А в образце.
Результаты титриметрического определения представляют в виде массового содержания или массовой доли в процентах вещества в пробе.
Вычисление содержания определяемого вещества проводят по нормальной концентрации титранта или чаще всего через титр соответствия. Этот способ вычисления удобен при массовых определениях одного и того же элемента в большом количестве проб. Вычислив однажды титр стандартного раствора по определяемому веществу Т(В/А), находят затем массу этого вещества простым умножением титра на затраченный объем раствора (формулы 2.6 и 2.8 а).
Так рассчитывают массовое содержание определяемого вещества А по способу отдельных навесок, можно рассчитать и массовую долю (в %) (формулы 2.10. и 2.17).
|
ω(A) |
c[(1/z)B]M(A)f |
экв (A)V(B)100 |
. |
2.17 |
|
1000 |
mобщ |
|||
Пример 2.6. Какова массовая доля (в %) KCl в навеске массой 0,1523 г, если на ее титрование израсходовали 20,10 см3 0,1015 М раствора AgNO3? Определение основано на реакции:
39
KCl + AgNO3 = AgCl↓ + KNO3 .
Молярная масса KCl равна 74,55 г/моль. Фактор эквивалентности равен 1.
|
ω(KCl) |
0,1015 74,55 |
20,10 100 |
99,86 % . |
|
|
1000 |
0,1523 |
|||
В методе пипетирования на титрование аликвоты Va(A) см3 раствора определяемого вещества А израсходовано V(B) cм3 стандартного раствора В с титром по определяемому веществу Т(В/А). Тогда в объеме Va(A) содержится Т(В/А)V(B)г определяемого вещества, а в мерной колбе Vобщ (А)
|
массу m(A) рассчитывают по формулам |
|||||||
|
m( A) |
T (B / A) V (B) Vобщ |
( А) |
, |
(2.18 а) |
|||
|
Va ( A) |
|||||||
|
m(A) |
c[(1/z)B] M(A) f экв (A) V(B) Vобщ (A) |
||||||
|
. |
(2.18 б) |
||||||
|
1000 |
Va (A) |
Массовую долю (в %) вещества в навеске mобщ рассчитывают по фор-
мулам 2.10 или 2.19.
|
ω(А( |
c[(1/z)B] M(A) fэкв (A) V(B) Vобщ (А)100 |
. |
(2.19) |
|
|
1000 |
Va (A) mобщ |
|||
Пример 2.7. В мерную колбу вместимостью 100,0 см3 перенесли 0,6504г образца щавелевой кислоты, растворили и довели объем раствора до метки. Пипеткой взяли 10,00 см3 полученного раствора, поместили в колбу для титрования и титровали 0,1026 М раствором NaOH, расход которого составил 9,85 см3. Определить массовую долю (в %) H2C2O4 · 2H2O в исследуемом образце.
Фактор эквивалентности щавелевой кислоты равен 1/2 , молярная масса составляет 132,066 г/моль, способ титрования – пипетирование. Рассчитывают массовую долю щавелевой кислоты по формуле 2.19.
|
ω |
0,1026 |
66,033 9,85 100,0 100 |
97,97 %. |
|
|
1000 10,00 0,6504 |
||||
В случае титрования по остатку (обратное титрование), когда используется два титрованных раствора с1 и с2, массу определяемого вещества в методе отдельных навесок вычисляют по формуле
|
m(A) |
(c |
1V1 |
c2V2 )M(A)f экв |
(A) |
. |
(2.20) |
|
1000 |
||||||
Соответственно массовую долю (в %) рассчитывают по формуле
|
ω(A) |
(c1V1 c2V2 |
) M(A) f экв |
(А)100 |
(2.21) |
|
|
1000 |
mобщ |
||||
Пример 2.8. К раствору 0,3850 г вещества, содержащему хлориды, прибавили 25,00 см3 0,1100 М раствора AgNO3. На титрование избытка AgNO3

40
затрачено 13,50 см3 0,1020 М раствора NH4NCS. Рассчитать массовую долю (в %) хлора в навеске вещества.
Определение основано на реакциях:
Cl– + Ag+ = AgCl↓,
Ag+ + NCS– = AgNCS↓.
|
Фактор эквивалентности хлорид-иона равен 1. |
|||||
|
( Cl ) |
[c(AgNO |
3 ) V(AgNO3 ) |
c(NH 4 NCS) V(NH 4 NCS)] M(Cl) 100 |
||
|
1000 |
mобщ |
||||
|
(0,1100 |
25,00 |
0,1020 13,50) 35,45 100 |
12,64 % . |
||
|
1000 |
0,3850 |
||||
При определении по методу пипетирования в обратном титровании расчет массовой доли (в %) проводят по формуле
|
ω(А( |
(с1V1 c2V2 ) M(A) fэкв (A) Vобщ (A) 100 |
|||
|
. |
(2.22) |
|||
|
1000 |
Va (A) |
mобщ |
||
|
Пример 2.9. 5,00 см3 раствора KClO3 |
поместили в мерную колбу емко- |
стью 100,0 см3, объем довели до метки. С помощью пипетки отобрали 10,00 см3 приготовленного раствора, перенесли в колбу для титрования, добавили по бюретке 20,00 см3 0,1200 н. раствора FeSO4, избыток оттитровали 10,15 см3 0,1105 н. раствора KMnO4. Рассчитать массовую долю (в %) KClO3 в растворе, если плотность этого раствора равна 1,040 г/см3.
Для определения использовали обратное титрование, основанное на реакциях:
ClO3– + 6Fe2+ + 6H+ = Cl– + 6Fe3+ + 3H2O,
5Fe2+ + MnO4– + 8H+ = 5Fe3+ + Mn2+ + 4H2O.
Фактор эквивалентности KClO3 по уравнению полуреакции
ClO3– + 6H+ + 6ē = Cl– + 3H2O
равен 1/6, молярная масса KClO3 равна 122,550 г/моль, молярная масса эквивалента – 22,425 г/моль.
|
Массовую долю (%) KClO3 |
находят по формуле 2.22, с учетом того, что |
||||||||||
|
массу навески рассчитывают по плотности и объему (Vконц) исследуемого |
|||||||||||
|
раствора (m = Vρ): |
|||||||||||
|
ω(KClO3 ) |
[c(FeSO4 ) V(FeSO4 ) c(1/5KMnO4 ) V(KMnO4 )] |
||||||||||
|
1000 |
|||||||||||
|
M(KClO3 ) f экв Vобщ 100 |
, |
||||||||||
|
Va (KClO3 ) Vконц ρ |
|||||||||||
|
ω(KClO3 ) |
(0,1200 |
20,00 |
0,1105 10,15) 22,425 100,0 100 |
5,52 % . |
|||||||
|
1000 |
10,00 |
5,00 |
1,040 |
||||||||
41
Формулы расчета массового содержания определяемого компонента приведены в табл. 2.2.
2.5.Кривые титрования
Впроцессе титрования изменяются равновесные концентрации определяемого вещества, титранта и продукта реакции. При этом пропорционально концентрациям веществ изменяются и свойства раствора. Например, при кислотно-основном титровании меняется рН раствора, при окислительновосстановительном титровании – окислительно-восстановительный потенциал системы. График зависимости параметра системы (рН, Е), связанного с концентрацией титруемого вещества, титранта или продукта, от состава раствора в процессе титрования называют кривой титрования.
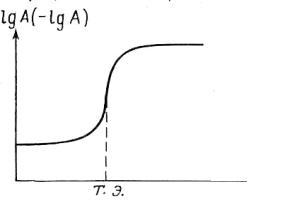
Кривые титрования помогают выбрать индикатор, оценить погрешность, наглядно проследить за ходом титрования. При построении кривых по оси ординат можно отложить логарифм концентрации или величину, пропорциональную этому логарифму, по оси абсцисс – объем добавленного титранта или степень оттитрованности титруемого вещества (обычно в процентах). Кривая титрования в общем виде представлена на рис. 2.2.
Рис. 2.2. Кривая титрования
А – концентрация определяемого компонента;
V – объем титранта;
Т.Э. – точка эквивалентности
V
Как видно из рис. (2.2), логарифмическая кривая имеет s-образную форму. На кривой имеются области плавного (до и после точки эквивалентности) и резкого (вблизи Т.Э.) изменения рассчитываемого или измеряемого параметра. Область резкого изменения (вертикальный участок кривой) называют скачком титрования. Если выразить степень оттитрованности в процентах, то скачок титрования начинается, когда оттитровано 99,9 % компонента и заканчивается, когда добавлено 100,1 % титранта. На величину скачка титрования влияет ряд факторов: константа равновесия реакции, концентрация веществ, температура и др.
При визуальном обнаружении точки эквивалентности пользуются индикаторами. Это вещества, окраска которых меняется при определенных значениях параметра. Индикатор выбирают так, чтобы его окраска изменялась в пределах установленного скачка титрования (не обязатель-
42
но в точке эквивалентности). При этом, если индикатор меняет свой цвет до Т.Э., возникает погрешность титрования со знаком минус, если изменяется цвет после Т.Э. – со знаком плюс. Очевидно, что для правильного выбора индикатора и оценки погрешности титрования необходимо построение кривой титрования.
Контрольные вопросы и задачи
1.В чем заключается сущность титриметрического анализа? Какой закон лежит в основе титриметрии?
2.Каким требованиям должны удовлетворять реакции, применяемые в титриметрическом анализе?
3.Что такое точка эквивалентности и как она фиксируется?
4.Что называют эквивалентном, эквивалентным числом, фактором эквивалентности?
5.Является ли молярная масса эквивалента вещества постоянной величиной? От чего зависит ее значение?
6.Определить фактор эквивалентности участников следующих реакций:
Na2CO3 + HCl = NaHCO3 + NaCl; Na2CO3 + 2HCl = NaCl + CO2 + H2O; H3PO4 + 2NaOH = Na2HPO4 + 2H2O;
2KMnO4 + 10FeSO4 + 8H2SO4 = 2MnSO4 + 5Fe2(SO4)3 + K2SO4 +8H2O; Ca(OH)2 + H2SO4 = CaSO4 + 2H2O.
7.Какой измерительной посудой измеряются точные объемы растворов в титриметрии?
8.С какой точностью взвешивают аналитические навески?
9.Почему бюретки и пипетки необходимо перед употреблением промыть тем раствором, которым их будут наполнять?
10.Что такое «первичный стандартный раствор», «вторичный стандартный раствор»?
11.Какие требования предъявляют к первичным стандартным веществам? 12.Что такое «фиксанал» или «стандарт-титр»?
13.Стандартизацию раствора HCl можно сделать по титрованному раствору NaOH и по х.ч. Na2B4O7 · 10H2O. Какой из этих способов обеспечит более высокую точность и почему?
14.Что такое титр раствора и как он связан с нормальной концентрацией раствора?
15.Что такое титр соответствия или титр по определяемому веществу? 16.В 400 см3 раствора серной кислоты содержится 4,9000 г безводной
H2SO4. Вычислите титр, молярную концентрацию и молярную концентрацию эквивалентов данного раствора. Ответ: 0,01226 г/см3; 0,1250 моль/дм3; 0,2500 моль/дм3.
43
17.В 250,0 см3 раствора гидроксида натрия содержится 10,00 г этого вещества. Чему равен титр этого раствора? Ответ: Т(NaOH) = 0,04000 г/см3.
18.Чему равны титры 0,0900 н. раствора H2SO4 по: а) Ba(OH)2; б) NH3? От-
вет: а) 0,007712 г/см3; б) 0,001593 г/см3.
19.Вычислить нормальную концентрацию раствора хлороводородной кислоты, если T(HCl/NaOH) = 0,005250 г/см3. Ответ: 0,1312 моль/дм3.
20.Сколько граммов КОН содержится в 10 см3 раствора, титр которого ра-
вен 0,004120 г/см3. Ответ: 0,04122 г.
21.Как проводятся титриметрические определения по способу пипетирования и по способу отдельных навесок?
22.Вычислить количество граммов Na2CO3 в растворе, на титрование которого израсходовано 22,00 см3 0,1200 М раствора HCl? Ответ: 0,1399 г.
23.Сколько граммов H2SO4 содержится в 5 дм3 раствора, если на титрование 25,00 см3 этого раствора израсходовано 22,50 см3 0,09500 М раствора КОН? Ответ: 20,97г.
24.Какую массу навески Na2CO3 нужно взять, чтобы на титрование ее требовалось 20 см3 0,1н. раствора H2SO4? Ответ: 0,11г.
25.Какому объему 1 М раствора эквивалентны 23,8 см3 0,2 М раствора HСl?
ГЛАВА 3. КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ (ПРОТОЛИТОМЕТРИЯ)
3.1. Сущность метода
Метод кислотно-основного титрования базируется на реакции обмена протонами между протолитами (кислотами и основаниями, по БренстедуЛоури) и в общем виде может быть представлен схемой
кислота1 + основание2 = основание1 + кислота2, НА + В = ВН+ + А–, Н3О+ + ОН– = Н2О.
Кислотно-основное титрование впервые было предложено в 1823 г. Гей-Люссаком.
Реакции кислотно-основного взаимодействия отвечают требованиям, предъявляемым к реакциям в титриметрии, в отношении их стехиометрии и скорости протекания. Связывание Н3О+ и ОН– в растворе происходит за время порядка 10-11с. Применение в качестве титрантов только сильных кислот (HCl, H2SO4) и оснований (КОН, NaOH) позволяет достичь достаточной полноты протекания реакции (Кр ≥ 1 · 108) при титровании не только сильных, но и слабых протолитов, константы диссоциации (Ка, Кв) ко-
44
торых не меньше 1 · 10-8. Фиксирование точки эквивалентности осуществляется вполне доступными методами, в основном, с помощью индикаторов.
В зависимости от титранта различают ацидиметрическое и алкалиметрическое титрование. Название происходит от латинских слов acidum
– кислота и alkali – щелочь.
Ацидиметрическое титрование (титрант – кислота) применяют для определения протолитов, проявляющих свойства оснований: сильных и слабых оснований, основных солей, солей слабых кислот, а также органических соединений, обладающих основными свойствами.
Алкалиметрическое титрование (титрант – щелочь) используют для определения сильных и слабых кислот, кислых солей, солей слабых оснований и органических соединений, обладающих кислотными свойствами.
При кислотно-основном титровании главным свойством системы, которое меняется пропорционально количеству оттитрованного протолита, является кислотность раствора, характеризующаяся величинами [H3O+] или рН. В зависимости от соотношения силы реагирующих протолитов точка эквивалентности может находится в кислой, нейтральной и щелочной средах. В соответствии с этим рассматривают три случая титрования:
1. Титрование сильной кислоты щелочью (или наоборот):
HCl + NaOH = NaCl + H2O,
H+ + OH– = H2O.
Образующаяся соль NaСl не подвергается гидролизу, и раствор будет иметь нейтральную реакцию. Следовательно, в данном случае точка эквивалентности находится в нейтральной среде (рН=7).
2. Титрование слабой кислоты щелочью:
CH3COOH + NaOH = CH3COONa + H2O.
В точке эквивалентности образуется ацетат натрия – соль слабой кислоты и сильного основания, которая гидролизуется:
CH3COONa + H2O = CH3COOH + NaOH,
CH3COO– + H2O = CH3COOH + OH–.
В растворе появляется избыток ионов ОН–, поэтому среда в точке эквивалентности щелочная (рН > 7).
3. Титрование слабого основания сильной кислотой:
NH4OH + HCl = NH4Cl + H2O.
Образующаяся в точке эквивалентности соль NH4Cl гидролизуется:
NH4Cl + H2O = NH4OH + HCl,
NH4+ + H2O = NH4OH + H+.
В результате накопления ионов H+ среда в точке эквивалентности становится кислой (рН < 7).

45
Таким образом, в двух последних случаях точка эквивалентности не совпадает с точкой нейтральности. (Поэтому не рекомендуется использовать старое название «метод нейтрализации»).
3.2. Индикаторы кислотно-основного титрования
Фиксирование точки эквивалентности при кислотно-основном титровании проводят с помощью индикаторов или инструментальными методами (потенциометрически, кондуктометрически и т.п.).
Индикаторы метода – кислотно-основные индикаторы – представляют собой слабые органические кислоты или основания, протонированные (кислотные) и непротонированные (основные), формы которых различаются по структуре и окраске. Существуют одноцветные (например, фенолфталеин) и двухцветные (например, метиловый оранжевый) индикаторы. Кислотно-основные индикаторы относятся к обратимым индикаторам.
В соответствии с ионно-хромофорной теорией изменение окраски индикатора связано с таутомерией органических молекул, содержащих хромофорные группы. Хромофоры, или носители цветности, содержат π- электроны. Наиболее известными хромофорами являются группы:
>С=О, >С=С<, >С=S<, >C=N—, —N=N—, —O—N=N—, —N=0, >C=C— C=C<, = = =. Последняя группа называется хиноидной. Соединения, содержащие хромофорные группы, имеют полосу поглощения в ультрафиолетовой или видимой областях спектра. Если молекулы поглощают свет в видимой части спектра, то вещество имеет определенную окраску.
Соединениям с хромофорными группами, содержащими подвижные π- электроны, в зависимости от распределения электронной плотности можно приписать несколько структур; предельные структуры называются ее таутомерами. На распределение электронной плотности влияет наличие аук-
сохромных групп (NH2–, –OH, –OCH3, –N(CH3)2, –COO и т.п.). Ауксохро-
мы связаны с ненасыщенным скелетом хромофора так, что положение двойных связей изменяется и поглощение света усиливается. Таким образом, ауксохромы сами не сообщают окраску индикатору, но обладают свойством усиливать действие хромофоров повышать интенсивность вызываемой ими окраски.
Рассмотрим индикатор метиловый оранжевый. В щелочной среде индикатор имеет желтую окраску (хромофор –N=N–). В кислой среде азот, содержащий неподеленную пару электронов, протонируется, при этом образуется хиноидная форма и окраска становится красной. В растворе в равновесии находятся обе таутомерные формы.
Метиловый оранжевый относится к классу азоиндикаторов (-N=N-), имеющих в кислой среде красную, а в щелочной среде желтую окраску. Сюда относится также метиловый красный и тропеолин О.
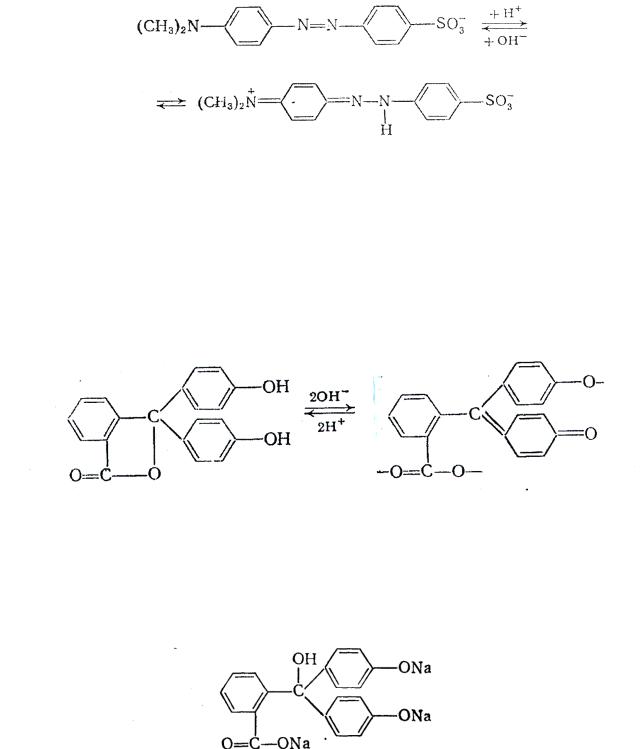
46
желтая форма (нейтральная или щелочная среда))
красная форма (кислая среда)
Существуют индикаторы, не содержащие хромофоров, но под влиянием рН среды их структура изменяется так, что хромофорные группы появляются. Это группа фталеинов. Например, фенолфталеин, тимолфталеин. Так, фенолфталеин в кислой среде бесцветен. В щелочной среде (рН 8-10) в результате перераспределения электронной плотности в его молекуле образуется хиноидная структура (хромофор), находящаяся в равновесии со своей таутомерной формой. Индикатор приобретает малиново-красную окраску:
|
бесцветная форма |
окрашенная форма |
Дальнейшее увеличение рН до 13-14 вызывает новую структурную перегруппировку, в результате чего образуются анионы трехзамещенной соли, лишенные хиноидной группы и потому бесцветные. Вследствие этого фенолфталеин обесцвечивается при действии большого избытка щелочи:
Кислотно-основные индикаторы, меняющие свой цвет в зависимости от концентрации ионов Н+ являются специфическими индикаторами на эти ионы. Известно большое число разных кислотно-основных индикаторов, сведения о некоторых из них приведены в табл. 3.1.

47
|
Таблица 3.1 |
|||||
|
Кислотно-основные индикаторы |
|||||
|
Область пере- |
Изменение окраски |
||||
|
Индикатор |
рКа |
||||
|
кислотная |
основная |
||||
|
хода рН |
|||||
|
форма |
форма |
||||
|
Метиловый фиолетовый |
0-1,8 |
– |
желтая |
фиолетовая |
|
|
Тимоловый синий |
1,2-2,8 |
1,65 |
красная |
желтая |
|
|
Метиловый оранжевый |
3,1-4,4 |
3,36 |
красная |
желтая |
|
|
Бромкрезоловый зеленый |
3,9-5,4 |
4,90 |
желтая |
синяя |
|
|
Метиловый красный |
4,4-6,2 |
5,00 |
красная |
желтая |
|
|
Бромтимоловый синий |
6,0-7,6 |
7,3 |
желтая |
синяя |
|
|
Феноловый красный |
6,4-8,2 |
8,00 |
желтая |
красная |
|
|
Тимоловый синий |
8,0-9,6 |
9,20 |
желтая |
синяя |
|
|
Фенолфталеин |
8,2-9,8 |
9,53 |
бесцветная |
красная |
|
|
Тимолфталеин |
9,3-10,5 |
9,6 |
бесцветная |
синяя |
|
|
Ализариновый желтый |
9,7-10,8 |
– |
желтая |
красная |
|
Равновесие в растворе кислотно-основного индикатора можно представить схематично следующим образом:
HInd + H2O Ind– + H3O+.
Константа равновесия этой реакции, являющаяся константой кислотности, иногда называемой константой индикатора, имеет выражение:
|
KInd |
[Ind |
] [H 3O ] |
. |
|
[HInd] |
|||
Из выражения константы индикатора получаем
|
[H 3O ] K Ind |
[HInd] |
, рН |
рКInd |
lg |
[HInd] |
. |
|
[Ind ] |
||||||
|
[Ind ] |
Так как обе формы индикатора окрашены различно, цвет индикатора при данных условиях рН среды зависит от соотношения [HInd]/[Ind–]. Человеческий глаз воспринимает окраску одной из форм, если ее концентрация превышает концентрацию другой формы примерно в 10 раз. Тогда в
|
интервале рН от pKInd |
lg |
10 |
до pK Ind |
lg |
1 |
, глаз будет видеть смешан- |
|
|
1 |
10 |
||||||
ную окраску обеих форм, а за пределами этого интервала – чистую окраску одной из форм. Этот интервал называют интервалом перехода окраски индикатора:
рН рКInd 1.
48
Очевидно что, если глаз улавливает окраску одной из форм на фоне другой при большем или меньшем соотношении, ∆рН будет другим. Чем меньше интервал, тем ценнее индикатор.
Середина области перехода окраски индикатора называется показателем индикатора рТ (при этом рН = рКInd).
Киндикаторам предъявляют ряд требований:
1.Индикатор должен быть интенсивно окрашен, чтобы окраска даже его небольшого количества была заметна для глаза. Большая концентрация индикатора может привести к расходу на него значительного количества (больше капли) титранта.
2.Переход окраски должен быть контрастным.
3.Область перехода окраски должна быть как можно уже.
Для сужения области перехода окраски и увеличения контрастности применяют смешанные индикаторы, которые составляют из индикатора и красителя или из двух индикаторов. Так, у индикатора метилового красного, переход цвета от красного и желтому (табл. 3.1) происходит в интервале почти двух единиц рН. Если к раствору добавить подходящее количество красителя метиленового синего, то переход от красно-фиолетовой окраски к зеленой наблюдается резко и отчетливо при рН = 5,3. Тот же метиловый красный в смеси с бромкрезоловым зеленым при рН = 5,1 дает отчетливый переход от красного цвета к зеленому.
На область перехода окраски индикатора (положение и интервал) влияют ионная сила, температура, посторонние вещества, растворитель, а также концентрация индикатора. Из посторонних веществ следует отметить влияние углекислого газа и веществ, образующих коллоидные системы. За счет углекислого газа рН водного раствора уменьшается, поэтому все индикаторы с рТ > 4 чувствительны к СО2. Если в растворе имеются коллоидные системы, например, белки, то наблюдается адсорбция индикатора на поверхности коллоидных частиц. Кроме того, происходит взаимодействие кислотных и основных групп белков и индикаторов, что приводит к ошибкам титрования.
3.3. Кривые титрования и выбор индикатора
Для правильного выбора индикатора изучают, как меняется рН раствора в процессе титрования. Значение рН можно рассчитать или измерить с помощью прибора рН-метра. По полученным данным строят кривую титрования, то есть график зависимости рН от объема прибавленного титранта (или от степени оттитрованности). В зависимости от определяемых компонентов различают три вида кривых титрования.
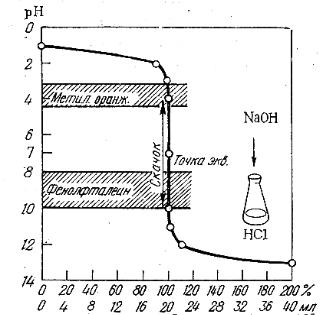
При титровании сильной кислоты щелочью (или наоборот), точка эквивалентности находится на линии нейтральности (рН = 7) и при концентрации вещества и титранта, равным 0,1 моль/дм3, скачок титрования находится в интервале рН от 4 до 10 (рис. 3.1).
49
В идеале для фиксирования точки эквивалентности подходит индикатор, у которого рТ совпадает с рН в момент эквивалентности, в данном случае можно использовать бромтимоловый синий (табл 3.1). Но можно применять также индикаторы, меняющие окраску в пределах рН от 4 до 10, ошибка титрования при этом будет незначительная. Так, с метиловым оранжевым (рТ = 3,4) раствор будет слегка недотитрован, с фенолфталеином (рТ = 9,5) – перетитрован.
Рис.3.1. Кривая титрования
0,1 М раствора HCl 0,1 М раствором NaOH
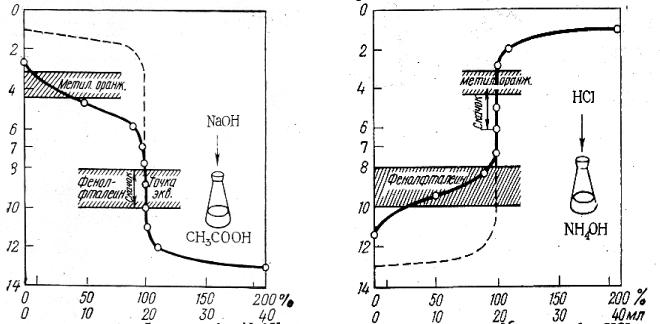
Кривая титрования слабой уксусной кислоты щелочью (0,1 М растворы) представлена на рис. 3.2, здесь скачок титрования охватывает интервал значения рН 7,76 – 10 и точка эквивалентности лежит в щелочной среде при рН 8,88. Для фиксирования точки эквивалентности подходит фенолфталеин.
Кривая титрования слабого основания (раствора аммиака) сильной кислотой (0,1 М растворы) приведена на рис. 3.3. Скачок титрования находится в пределах рН 4-6,24, точка эквивалентности лежит в кислой среде при рН 5,12 и может быть зафиксирована метиловым оранжевым.
Таким образом, кислотно-основное титрование можно проводить с индикатором, интервал перехода окраски которого находится в области скачка рН на кривой титрования.
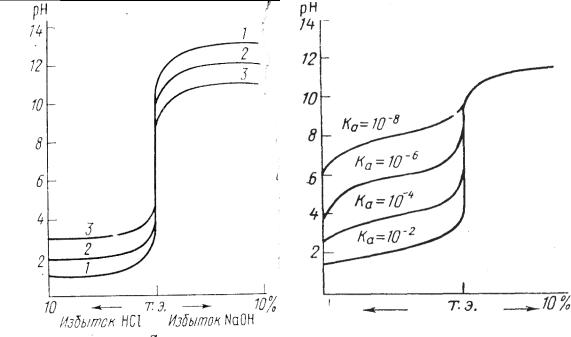
Чем больше скачок титрования, тем отчетливее и с меньшей погрешностью можно зафиксировать момент эквивалентности. Величина скачка титрования зависит от концентрации, а для слабых протолитов и от их констант кислотности и основности (Ка, Кв). Чем сильнее электролит и выше его концентрация, тем больше скачок титрования (рис. 3.4 и 3.5).
50
Слабые протолиты с К ≤ 1,0 · 10-8 вообще не дают скачка на кривой титрования и прямым титрованием в водном растворе не определяются.
При титровании растворов высоких концентраций (с ≥ 1,00 М) возникает погрешность вследствие того, что ошибка титрования в одну каплю составляет уже значительную величину. Оптимальный интервал концентраций растворов при титровании 0,2-0,02 М. Правильное определение конечной точки титрования зависит не только от выбора индикатора, но и от принятого в работе порядка титрования. Необходимо учитывать визуальное восприятие цвета раствора человеческим глазом. Титрование можно проводить «от кислоты к щелочи» и на оборот. При титровании «от кислоты к щелочи» с метиловым оранжевым розовая окраска индикатора от избыточной капли щелочи должна перейти в желтую. Но такое изменение окраски гораздо хуже улавливается глазом, чем переход от желтой в розовую. Поэтому с метиловым оранжевым (или метиловым красным) титруют «от щелочи к кислоте». С фенолфталеином удобнее титровать «от кислоты к щелочи», так как при этом раствор из бесцветного становится малиновым.
Рис. 3.2. Кривая титрования 0,1 М раствора CH3COOH 0,1 М раствором NaOH
Рис. 3.3. Кривая титрования 0,1 М раствора NH4OH 0,1 М раствором
HCl
Кроме того, правильное фиксирование точки эквивалентности зависит от количества прибавленного индикатора. Чем больше реактива, тем ярче окраска раствора, но тем труднее заметить изменение окраски. Для установления момента эквивалентности имеет значение не столько яркость
51
окраски раствора, сколько четкость ее изменения. Следует помнить, что индикаторы, применяемые в кислотно-основном титровании, сами являются слабыми кислотами или основаниями и при титровании часть стандартного раствора расходуется на титрование индикатора. Опытным путем установлено, что на 10-15 см3 анализируемого раствора достаточно одной капли индикатора, а для 25 см3 – две. Причем при параллельных определениях следует брать одно и то же количество индикатора.
|
10 % |
|
|
Рис. 3.4. Зависимость скачка на |
Рис. 3.5. Зависимость величины |
|
кривой титрования от концен- |
скачка на кривой титрования |
|
трации раствора: |
от силы протолитов |
|
1 – 1,0 М; 2 – 0,1М; 3 – 0,01 М |
На изменение окраски индикатора также влияет присутствие нейтральных электролитов, растворители, температура.
Для учета погрешностей, возникающих при титровании с данным индикатором рекомендуется использовать контрольный раствор (называемый также «холостым» или «свидетелем»). «Холостым» называют титрование раствора, идентичного с анализируемым (по объему, количеству индикатора и т.п.), но не содержащего определяемого вещества. Объем титранта, пошедший на контрольное титрование, можно вычесть из результатов основного титрования, тем самым устранив одну из погрешностей титриметрического определения.
Очень часто в качестве «свидетеля» используют дистиллированную воду, помещаемую в колбу для титрования в количестве равном объему жид-
![]()
52
кости в конце титрования, куда добавляют индикатор и 1-2 капли титранта до изменения цвета индикатора. Приготовленный таким образом «свидетель» используют в качестве образца при титровании, добиваясь, чтобы окраска анализируемого раствора и «свидетеля» была одинакова.
3.4.Титранты метода
Вкачестве титрантов чаще всего используют вторичные стандартные растворы HCl и NaOH. Их готовят, в основном, из концентрированных растворов разбавлением приблизительно до требуемой концентрации с последующей стандартизацией по первичным стандартным веществам.
3.4.1. Приготовление и стандартизация растворов кислот и щелочей
Стандартные растворы кислот и щелочей обычно готовят с молярной концентрацией эквивалентов вещества с[(1/z)B] от 0,05 до 0,1 реже 1 моль/дм3. С помощью ареометра (денсиметра) определяют плотность концентрированной кислоты или щелочи. Ареометр (рис. 3.6) представляет собой герметически закрытый цилиндрический сосуд с нанесенными на нем метками. В нижней части помещен груз (дробь), благодаря чему ареометр, погруженный в жидкость, поддерживается в вертикальном положении. Для определения плотности концентрированную кислоту или щелочь наливают в сухой стеклянный цилиндр объемом 200-250 см3. Правильно подобранный ареометр не должен касаться дна и стенок цилиндра. Отсчет показаний ведут по шкале ареометра. Деление шкалы, совпадающее с уровнем жидкости, показывает плотность раствора ρ (г/см3). По справочным таблицам определяют соответствующую плотности массовую долю (в %) или молярную концентрацию раствора. Если молярная концентрация не указана, ее рассчитывают по формуле (2.12 а), затем переводят в нормальную (если z > 1, например в H2SO4).
Рис. 3.6. Ареометр
53
Требуемый объем исходного концентрированного раствора кислоты или щелочи, необходимый для приготовления раствора заданной концентрации, рассчитывают по формуле 2.13 в.
Рассчитанный объем концентрированной кислоты или щелочи измеряют с помощью мерного цилиндра или градуированной пробирки, переносят в чистую сухую склянку, прибавляют соответствующий объем дистиллированной воды, перемешивают и закрывают пробкой. Таким образом получают раствор приблизительной концентрации.
Точную концентрацию приготовленной кислоты устанавливают с помощью первичных стандартных веществ, имеющих в растворе щелочную реакцию. К ним относятся безводные карбонат или гидрокарбонат натрия Na2CO3, NaHCO3 и тетраборат натрия (бура) Na2B4O7 · 10H2O. Наиболее удобен тетраборат натрия, так как путем перекристаллизации при 600С и высушивании на воздухе эту соль легко получить химически чистой, точно соответствующей формуле Na2B4O7 · 10H2O. Это вещество устойчиво, имеет достаточно большую молярную массу эквивалента (190,7 г/моль), что отвечает требованиям для установочных (ПСВ) веществ.
Водный раствор тетрабората натрия вследствие гидролиза имеет щелочную реакцию и потому может быть оттитрован кислотой.
Na2B4O7 + 2HCl + 5H2O = 2NaCl + 4H3BO3.
В результате реакции накапливается ортоборная кислота. Следовательно, рН раствора в точке эквивалентности слабокислая и для титрования можно использовать индикатор метиловый оранжевый (или метиловый красный).
Стандартизацию кислоты по тетраборату натрия можно проводить как методом пипетирования, так и по отдельным навескам. Большая масса эквивалента позволяет воспользоваться методом отдельных навесок.
Массу навески тетрабората натрия рассчитывают по формуле 2.8 а (см. пример 2.2) исходя из того, что на ее титрование должно расходоваться около 20,00 см3 0,1 М раствора HCl (если готовили раствор кислоты такой концентрации).
Рассчитанную массу навески взвешивают предварительно на технических (или аптечных) весах, затем переносят в бюкс и находят точную массу бюкса с навеской на аналитических весах. Результаты взвешивания записывают в лабораторный журнал. Навеску переносят в коническую колбу для титрования объемом 200-250 см3 . Взвешивают бюкс с оставшимися крупинками и вновь записывают массу в журнал. По разнице двух взвешиваний на аналитических весах находят точную массу тетрабората натрия.
В колбу с навеской добавляют 20-25 см3 горячей дистиллированной воды (бура плохо растворяется в холодной воде), смывая частицы вещества со стенок колбы. Содержимое колбы перемешивают осторожными враща-
54
тельными движениями до растворения вещества. Затем раствор охлаждают до комнатной температуры и добавляют 1-2 капли индикатора метилового оранжевого.
Установленную в штативе чистую бюретку ополаскивают 2 раза приготовленным раствором кислоты, затем наполняют ее почти доверху. Далее, подставив стаканчик под бюретку и приоткрыв зажим, заполняют нижний конец бюретки (носик) так, чтобы в ней не осталось пузырьков воздуха (см. рис.1.11). Нижний мениск раствора кислоты в бюретке должен находиться на нулевом делении (рис. 1.13).
Колбу с раствором тетрабората натрия ставят на белую фарфоровую пластинку или на лист бумаги и осторожно титруют, непрерывно перемешивая раствор в колбе вращательными движениями (рис. 1.14). Необходимо уловить момент, когда от одной капли кислоты раствор в колбе изменит окраску из желтой на оранжевую. Для более точного фиксирования точки эквивалентности рекомендуется титровать по «свидетелю» (см. раздел 3.3).
Титрование первой навески следует проводить очень медленно, чтобы не перетитровать. Вторую и последующие навески можно титровать быстрее. В случае возникновения сомнения в изменении окраски, делают отсчет по бюретке и добавляют еще 1 каплю кислоты. Если при этом раствор порозовеет, значит, титрование было закончено в точке эквивалентности (учитывать объем лишней капли кислоты не следует). Зафиксированный по бюретке объем с точностью до сотых долей см3 (последний знак определяется на глаз), записывают в лабораторный журнал (например, V(HCl)
= 19,85 см3).
Расчет концентрации кислоты проводят до четвертой значащей цифры для каждой навески отдельно. Среднее значение находят по двум-трем сходящимся результатам (отличие может быть только в третьей значащей цифре). Кроме концентрации кислоты с такой же точностью рассчитыва-
ют ее поправочный коэффициент (K = cпракт/стеор). Приготовленный вторичный стандартный раствор кислоты (титрант) помещают в чистую
сухую склянку, закрывают пробкой, наклеивают этикетку с обозначением раствора и его концентрации. Раствора HCl довольно устойчив.
Стандартизацию аналогично приготовленного раствора щелочи проводят по ПСВ: щавелевой кислоте H2C2O4·2H2O, янтарной кислоте H2C4H4O4, бензойной кислоте C6H5COOH и другим органическим кислотам. При взаимодействии слабых органических кислот со щелочью образуется гидролизирующиеся соли, создающие в точке эквивалентности щелочную среду, которую можно зафиксировать индикатором фенолфталеином. Например:
H2C2O4 + 2NaOH = Na2C2O4 + 2H2O,
C6H5COOH + NaOH = C6H5COONa + H2O.
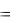
55
Случай стандартизации раствора NaOH по щавелевой кислоте методом пипетирования приведен в примере 2.1. Там же даны расчеты массы навески и концентрации кислоты и щелочи.
Техника работы по методу пипетирования несколько отличается от приема отдельных навесок. Рассчитанную массу навески также вначале взвешивают на аптечных (или технических) весах, помещают в бюкс, взвешивают на аналитических весах, затем навеску аккуратно переносят через сухую воронку в мерную колбу, вновь взвешивают бюкс (уже без навески). По разнице масс первого и второго взвешивания находят массу навески щавелевой кислоты. Остатки щавелевой кислоты с воронки тщательно смывают в ту же колбу дистиллированной водой, колбу наполняют водой до половины ее объема и растворяют навеску. Затем доводят объем колбы водой до метки по нижнему мениску жидкости, добавляя последние капли пипеткой (рис. 1.9). Мерную колбу закрывают пробкой и перемешивают раствор, переворачивая колбу несколько раз.
По точной массе навески щавелевой кислоты и объему колбы рассчитывают фактическую нормальную концентрацию ее раствора.
Мерную пипетку, с помощью которой отбирают аликвотные (равные) объемы раствора, несколько раз промывают приготовленным раствором щавелевой кислоты. Затем набирают пипеткой раствор до метки (см. рис. 1.16) по нижнему мениску жидкости и переносят в чистую коническую колбу для титрования. Добавляют 2-3 капли фенолфталеина и титруют приготовленным раствором щелочи до появления бледно-малиновой окраски, не исчезающей при взбалтывании в течение 30 с.
Проводят не менее трех титрований, объемы щелочи при этом не должны отличаться более чем на 0,1 см3. Из таких сходящихся результатов
__
(промахи отбрасывают) находят среднее значение V . Затем рассчитывают точную концентрацию приготовленного раствора щелочи:
|
c(NaOH) |
c(H |
2C2O4 ) V(H 2C2O4 |
) |
. |
|
__ |
||||
|
V (NaOH) |
Стандартный раствор щелочи хранят в стеклянных сосудах, закрытых резиновой пробкой или в пластмассовых бутылях. Гидроксид натрия поглощает СО2 из атмосферы, поэтому его раствор предохраняют от попадания углекислоты с помощью трубочки, заполненной СаО или натронной известью, вставленной в пробку.
Имея стандартные растворы HCl и NaOH можно, определить с точностью до 0,1 % массу веществ, прямо или косвенно участвующих в протолитических реакциях. Если протолит сильный или слабый, но имеет константу равновесия больше, чем 1 · 10-8, то его количество можно определить прямым титрованием. Если протолит слабый (К < 1 · 10-8), то его

56
определение возможно косвенными методами: обратным титрованием (по остатку) или титрованием заместителя.
3.5. Примеры определений методом кислотно-основного титрования
Метод кислотно-основного титрования находит широкое применение при оценке качества различных групп потребительских товаров и особенно пищевых продуктов. Так, этим методом определяют общую (титруемую) кислотность, обусловленную содержанием свободных кислот и кислых солей в сырье и готовой продукции: муке, дрожжах, крахмале, макаронных и кондитерских изделиях, хлебе, безалкогольных напитках, молоке, квашеной капусте и т.д. Кислотно-основное титрование используют при анализе соды NaHCO3, уксусной, лимонной и других кислот, солей аммония, а также при определении азота в органических соединениях по методу Кьельдаля.
3.5.1. Определение массовой доли (%) NaHCO3 в твердом образце соды
Гидрокарбонат натрия при растворении в воде дает щелочную реакцию и может быть оттитрован раствором хлороводородной кислоты по прямому титрованию:
NaHCO3 + HCl = NaCl + H2CO3.
Слабокислую среду в точке эквивалентности (H2CO3) можно зафиксировать индикатором метиловым оранжевым.
Фактор эквивалентности NaHCO3 равен 1, молярная масса эквивалента составляет 84,0 г/моль. Если концентрация хлороводородной кислоты равна 0,1 моль/дм3, тогда при титровании по способу пипетирования и объеме мерной колбы 100,0 см3 масса навески соли составляет примерно 0,8 г.
|
m(NaHCO 3 ) |
0,1 84,0 100,0 |
0,84г 0,8 г. |
|
|
1000 |
|||
Примерно 0,8 г исследуемой соли отвешивают на аптечных весах, переносят в бюкс и взвешивают на аналитических весах. Содержимое бюкса через сухую воронку переносят в мерную колбу. Бюкс с остатками соли взвешивают на тех же аналитических весах. В лабораторном журнале делают записи, например:
|
масса бюкса с навеской |
21,91350 |
г |
|
масса пустого бюкса |
21,11445 |
г |
|
масса навески |
0,79905 |
г |
По разнице массы бюкса с навеской и без навески рассчитывают точную массу навески соли.
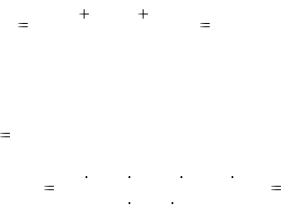
57
Дистиллированной водой смывают остатки соли с воронки в мерную колбу и заполняют ее до половины водой. Содержимое колбы перемешивают плавными круговыми движениями до полного растворения крупинок соли, затем доливают водой до метки по нижнему краю мениска, добавляя последние капли воды с помощью пипетки. Колбу закрывают пробкой и тщательно перемешивают раствор, неоднократно переворачивая колбу.
Мерную пипетку вместимостью 20,00 см3 ополаскивают приготовленным раствором соли. С помощью этой пипетки переносят аликвоту (Va) раствора соли в коническую колбу для титрования, добавляют 1 – 2 капли метилового оранжевого. Бюретку промывают 0,1 М раствором хлороводородной кислоты и заполняют ее до нулевой отметки. Титруют раствор гидрокарбоната натрия до перехода окраски от желтой в оранжевую. Можно приготовить два «свидетеля» – один с желтой окраской (без кислоты), другой с оранжевой (с 1 каплей кислоты). Так легче наблюдать резкий переход окраски при титровании.
Титрование проводят до трех сходящихся результатов. Перед каждым титрованием объем кислоты в бюретке доводят до нуля. Результаты титрования записывают в лабораторный журнал, например:
V1 = 19,50 см3 V2 = 19,00 см3 V3 = 18,90 см3 V4 = 18,95 см3
__
Средний объем V рассчитывают из трех последних значений объемов, считая первый результат «промахом».
|
__ |
19,00 |
18,90 |
18,95 |
18,95 см3 . |
|
V |
||||
|
3 |
||||
Значение среднего объема HCl используют для расчета массовой доли (в %) NaHCO3 в исследуемом образце.
|
__ |
||||||||
|
ω(NaHCO3 |
) |
c(HCl) M(NaHCO3 ) V (HCl) Vобщ (NaHCO3 |
) 100 |
, |
||||
|
1000 |
Va |
m |
||||||
|
ω(NaHCO3 |
) |
0,1 84,0 |
18,95 100,0 100 |
99,61% . |
||||
|
1000 |
20,0 0,79905 |
|||||||
3.5.2. Определение массовой доли (%) уксусной кислоты в растворе
Уксусная кислоты имеет константу кислотности Ка = 1,74 ·10-5 и может быть оттитрована щелочью по прямому титрованию:
CH3COOH + NaOH = CH3COONa + H2O.
В точке эквивалентности образуется гидролизирующаяся соль ацетата натрия, создающая щелочную среду. Следовательно, титрование можно проводить с индикатором фенолфталеином.
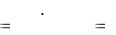
58
Однако следует учесть возникновение погрешностей данного определения. Титрование слабых кислот, какой является СН3СООН, происходит в присутствии угольной кислоты, находящейся в их водных растворах и в дистиллированной воде, а также образующейся при нейтрализации карбонатов, содержащихся в растворе NaOH.
Дистиллированная вода, не защищенная от действия диоксида углерода воздуха, имеет кислую реакцию (рН = 5,7). Для нейтрализации угольной кислоты с фенолфталеином затрачивается значительное количество раствора HCl. Для устранения влияния на результаты титрования угольной кислоты следует применять ряд особых мер: использовать свежеперегнанную дистиллированную воду, готовить стандартный раствор NaОН в условиях, исключающих попадание в него CO2, вблизи точки эквивалентности кипятить титруемый раствор для разрушения H2CO3, (с последующим охлаждением перед продолжением титрования).
При подготовке исследуемой пробы уксусной кислоты к анализу первоначально необходимо узнать приблизительную исходную концентрацию кислоты. Для этого с помощью ареометра определяют плотность раствора ρ, допустим, она составляет 1,015 г/см3. По справочным таблицам находят соответствующее плотности значение массовой доли (%) и молярной концентрации. Вышеуказанной плотности соответствует ω = 11,7 % и с(СН3СООН) = 1,98 моль/дм3. Если концентрация титранта NaOH равна 0,1 моль/дм3, то очевидно, что концентрация кислоты значительно выше и необходимо ее разбавить. С этой целью рассчитывают объем концентрированного раствора (Vконц) кислоты, необходимый для приготовления примерно 0,1 М раствора объемом, допустим, 100,0 см3 по формуле 2.13 в.
|
V |
0,1 100,0 |
5,05 см3 . |
|
|
конц |
|||
|
1,98 |
|||
Разводят исходный раствор по объему, для чего 5,00 см3 (Vконц) анализируемого раствора с помощью измерительной пипетки переносят в мер-
ную колбу вместимостью 100,0 см3 (Vобщ), доводят объем водой до метки, закрывают пробкой и хорошо перемешивают. Точную концентрацию раствора кислоты устанавливают титрованием 0,1 М раствором NaOH по способу пипетирования.
Методика определения. В колбу для титрования с помощью измерительной пипетки переносят 10,00 см3 разбавленного раствора уксусной кислоты, добавляют 1-2 капли фенолфталенина и титруют 0,1 М раствором NaOH до появления устойчивой в течение 30 с бледно-малиновой окраски. Титрование проводят до трех сходящихся результатов, например:
__
V1 = 9,90 см3; V2 = 9,80 см3; V3 = 9,85 см3; V = 9,85 см3
Массу навески концентрированного исходного образца уксусной кислоты находят по формуле m = Vконц · ρ. Рассчитывают массовую долю (%) СН3СООН в образце. М(СН3СООН) = 60 г/моль.
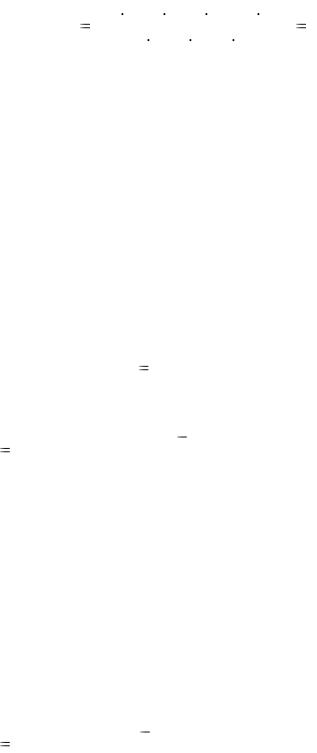
|
59 |
|||||||
|
ω(CH |
3COOH) |
0,1 60,0 |
9,85 100,0 100 |
11,64 % . |
|||
|
1000 10,0 |
5,00 |
1,015 |
|||||
3.5.3. Определение солей аммония и азота в органических соединениях по Кьельдалю
Определение иона аммония методом прямого титрования невозможно, так как NH4+ представляет собой очень слабую кислоту [К(NH4+) = 0,6 · 10-9]. Определяют соли аммония обычно методами обратного титрования или титрования по замещению.
В одном из методов обратного титрования к анализируемому раствору добавляют избыток точно измеренного титрованного раствора NaOH и нагревают до полного удаления NH3 из раствора (иначе аммиак титруется вместе со щелочью), после чего оставшееся количество щелочи определяют титриметрически с метиловым оранжевым.
|
t0 |
||||
|
NH4Cl + NaOH NH3 + H2O + NaCl, |
||||
|
NaOH + HCl = NaCl + H2O. |
||||
|
Массу аммиака в соли аммония рассчитывают по формуле |
||||
|
m(NH |
) |
[c(NaOH) V(NaOH) c(HCl) V(HCl)] M(NH3 ) |
. |
|
|
3 |
1000 |
|||
В другом методе обратного титрования к анализируемому раствору соли аммония добавляют избыток щелочи и выделившийся аммиак отгоняют в определенный заведомо избыточный объем титрованного раствора кислоты. Количество кислоты, оставшейся в растворе после взаимодействия с аммиаком, определяют титрованием щелочью по метиловому оранжевому. Применение фенолфталеина недопустимо, так как в реакцию могут вступить ионы аммония, присутствующие в растворе:
NH4Cl + NaOH = NH3 · H2O + NaCl,
NH3 + HCl = NH4Cl,
HCl + NaOH = NaCl + H2O.
Результат анализа рассчитывают по формуле:
|
m(NH |
) |
[c(HCl) V(HCl) c(NaOH) V(NaOH)] M(NH3 ) |
. |
|
|
3 |
1000 |
|||
Отгонка аммиака используется в широко известном методе определения азота в органических соединениях по Кьельдалю. В простейшем варианте этого метода пробу обрабатывают при нагревании концентрированной серной кислотой в присутствии катализатора, в результате чего органические соединения окисляются до СО2 и Н2О, а азот переходит в NH4HSO4. После охлаждения к остатку добавляют раствор щелочи и отгоняют NH3 (рис. 3.7) в отмеренный объем титрованного раствора кислоты, а
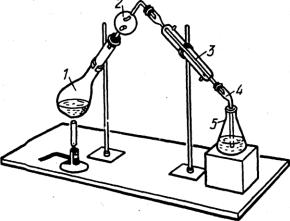
60
затем определяют избыток кислоты, не вошедшей в реакцию с аммиаком, и рассчитывают массу азота в пробе по формуле обратного титрования. Методом Кьельдаля можно определить азот в аминах, аминокислотах, алкалоидах, белках и многих других азотсодержащих соединениях. Этим методом определяют белок во всех белоксодержащих пищевых продуктах.
1. Колба Кьельдаля
2. Ловушка
3. Холодильник
4. Аллонж
5. Приемник
Рис. 3.7. Прибор для отгонки аммиака
Контрольные вопросы:
1.Какие из нижеперечисленных соединений можно определить мето-
дом кислотно-основного титрования: HNO3, NaNO3, NH4NO3, CaCl2, HCl, NH4Cl, Ba(OH)2, Na2CO3, H2SO4, KOH, H3PO4?
2.В каких случаях точка эквиваленнтости лежит при рН = 7 , при рН < 7, при рН > 7?
3.В каких случаях можно достаточно точно оттитровать относительно слабые протолиты?
4.Константа ионизации фенола Ка = 1·10-10. Возможно ли определение этой кислоты методом прямого кислотно-основного титрования?
5.Какие факторы или характеристики влияют на положение точки эквивалентности на кривой титрования, а также на величину и положение скачка титрования?
6.В чем заключается сущность ионно-хромофорной теории индикаторов? Что такое хромофоры и ауксохромы?
7.Что такое интервал перехода окраски индикатора и показатель титрования индикатора? Приведите значение показателя титрования для фенолфталеина и метилового оранжевого.
8.Почему не следует брать много индикатора при титровании?
9.Что такое индикаторная ошибка титрования и за счет чего она возникает?

61
10.Что такое «свидетель» и для чего его применяют при титровании? 11.Какой из индикаторов (метиловый оранжевый, фенолфталеин,
бромтиловый синий) с наименьшей погрешностью позволяет фиксировать точку эквивалентности при титровании: С6Н5СООН + NaOH; Na2CO3 + 2HCl; HNO3 + NaOH; NH3 · H2O + HCl?
12.Как, не прибегая к вычислениям, ориентировочно решить, какой индикатор выбрать для данного кислотно-основного титрования?
13.Какая методика – прямое или обратное титрование – используется при определении содержания следующих веществ: NH3 · H2O, Na2CO3, CH3COOH, NH4Cl, CaCO3? Составить соответствующие уравнения реакций.
14.Почему титр раствора NaOH нельзя найти по точной навеске твердого образца или концентрированного раствора?
15.Какие первичные стандартные вещества (ПСВ) используют для стандартизации: а) щелочи; б) кислоты?
16.По какой формуле рассчитывают массу навески ПСВ: а) по методу отдельных навесок; б) по методу пипетирования?
ГЛАВА 4. ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ ТИТРОВАНИЕ (РЕДОКСИМЕРИЯ)
4.1.Общая характеристика редокс-методов
Вредоксиметрии используют реакции окисления-восстановления. В основе метода лежит изменение потенциала окислительно-восстановительной системы при изменении соотношения концентраций окисленной и восстановленной форм в процессе титрования. В реакции участвуют две редокссистемы – титруемого вещества и титранта:
Ох1 + Red2 = Red1 + Ох2
Полнота протекания реакции зависит от разности стандартных потенциалов редокс-пар и константы равновесия реакции К:
Е = Е0(Ох1/Red2) – E0(Ox2/Red2),
|
lg K |
E n |
или |
K 10 |
E n |
, |
|
|
0,059 |
0,059 |
|||||
где n – число перемещаемых электронов, равное наименьшему кратному в полуреакциях.
Константа равновесия реакции тем больше, чем больше разность потенциалов. В этом заключается одна из основных причин того, что при титровании восстановителей предпочитаются титранты с высоким редокспотенциалом, а при титровании окислителей – восстановители с низким значением Е0. Кроме того, насколько более высок стандартный потенциал
![]()
62
используемой как окислитель редокс-пары, настолько больше восстановителей, которые можно оттитровать и определить с ее помощью. Так, стандартный потенциал окислителя KMnO4 в кислой среде (Е0(MnO4-, H+/Mn2+ = 1,51 В)) позволяет с достаточной полнотой протекания реакции оттитровать многие восстановители, например, соли железа (II):
E0(Fe3+/Fe2+) = 0,77 В,
5Fe2+ + MnO4– + 8H+ = 5Fe3+ + Mn2+ + 4H2O, ΔE = 1,51 – 0,77 = 0,74 В,
|
lg K |
0,74 |
5 |
64, К 1 1064 . |
|
|
0,059 |
||||
Разность потенциалов влияет и на величину скачка на кривой титрова-
ния, которую строят в координатах Е – V (титранта). Чем больше Е, тем больше скачок титрования и тем точнее титриметрическое определение.
Вотличие от реакций кислотно-основного титрования, окислительновосстановительные реакции протекают значительно сложнее. Во многих реакциях взаимодействуют не только окислители и восстановители, но и другие вещества (кислоты, щелочи). Часто реакции протекают в несколько стадий, причем каждая из них проходит с различной скоростью. Скорость окислительно-восстановительных реакций ниже скорости ионообменных реакций. Учитывая эти особенности протекания окислительно-восстановительных реакций, необходимо создать условия, при которых они могут удовлетворять требованиям титриметрии. Так, необходимую скорость реакций часто увеличивают искусственно: повышением температуры, концентрации реагирующих веществ, изменением рН раствора и применением катализатора.
Взависимости от применяемого титранта-окислителя редокс-методы
делят на перманганатометрию (KMnO4), иодометрию (J2), броматометрию (KBrО3), дихроматометрию (К2Cr2O7) и другие. В качестве титрантоввосстановителей применяют Na2S2O3, FeSO4, H2C2O4, Na3AsO3.
Для фиксирования точки эквивалентности в редоксиметрическом титровании используют: 1) исчезновение или появление окраски титранта или титруемого вещества; 2) окислительно-восстановительные и специфические индикаторы; 3) инструментальные методы.
Так, при титровании раствором KMnO4 с концентрацией не менее 0,02М раствор окрашивается в розовый цвет при введении одной лишней капли титранта.
Специфические индикаторы – это вещества, которые образуют интенсивно окрашенное соединение с одним из компонентов окислительновосстановительной системы. Например, при титровании иода используют крахмал, образующий темно-синее соединение с J3– – ионами.
63
Окислительно-восстановительные (редокс) индикаторы – это со-
единения, в основном органические, способные к окислению или восстановлению, причем их окисленная и восстановленная формы имеют разную окраску. Например, в качестве редокс-индикаторов используют дифениламин (см. качественную реакцию на нитрат-ион) и его производные. Редокс-индикаторы, как и рН-индикаторы, имеют интервал Е перехода окраски и подбираются для титрования по значению Е в точке эквивалентности или по скачку на кривой титрования.
4.2. Перманганатометрия
Перганатометрический метод анализа основан на реакции перманганата калия с восстановителями преимущественно в кислой среде:
MnO4– + 8H+ + 5ē = Mn2+ + H2O, Е0 = 1,51 В.
В слабокислых, нейтральных и щелочных растворах восстановление происходит до оксида марганца (IV), малорастворимого соединения бурого цвета:
|
MnO4– + 4H+ + 3ē = MnO2 ↓ + 2H2O, |
E0 |
= 1,69 В, |
|
MnO4– + 2H2O + 3ē = MnO2↓ + 4OH–, |
E0 |
= 0,60 В. |
В процессе титрования анализируемого раствора в кислой среде мали- ново-фиолетовая окраска перманганата калия обесцвечивается (ионы Mn2+ бесцветны). Добавление одной избыточной капли KMnO4 после достижения эквивалентности при титровании позволяет зафиксировать этот момент по появлению бледно-малиновой окраски раствора. Таким образом, не требуется дополнительный индикатор. В случае образования бурого осадка MnO2 фиксирование точки эквивалентности затрудняется.
Перманганат калия в кислой среде является сильным окислителем (Е0 =
1,51 В), и его применяют для определения многих восстановителей: ионов металлов (Fe2+, Sn2+, Cu+, Mn2+, Sb3+ и других), неметаллов (S2-, J–, Br–, SO32-,
S2O32-, CN–, NO2–, AsO33-, C2O42-), пероксида водорода. Метод перманганатометрии может быть использован для количественного определения не только восстановителей, но и окислителей. Окислители восстанавливают титрованным раствором щавелевой кислоты или арсенита натрия и избыток восстановителя оттитровывают перманганатом калия.
По заместительному титрованию возможно перманганатометрическое определение ионов, образующих малорастворимые оксалаты (Ca, Mg, Zn, Ba, Pb, Ag и др), которые отделяют от раствора, растворяют в кислоте и затем оттитровывают щавелевую кислоту перманганатом калия
Анализ некоторых органических соединений проводят в сильнощелочной среде:
MnO4– + ē = MnO42-, E0 = 0,56 В.
Органические соединения при этом обычно окисляются до карбонат-иона. По окончании реакции окисления избыток KMnO4 подкисляют и титруют стандартным раствором железа (II) или другого подходящего восстановителя. Таким образом опре-
64
деляют метиловый спирт, муравьиную, винную, лимонную кислоты и другие органические соединения.
Условия титрования в кислой среде. Процесс окисления неорганиче-
ских и органических веществ раствором перманганата калия довольно сложен. На различных стадиях протекания реакции образуются соединения марганца разной степени окисления Mn2+, Mn3+, Mn (IV), Mn (VI), причем, каждая ступень реакции проходит с различной скоростью. Для того чтобы все промежуточные стадии восстановления Mn (VII) в Mn2+ были количественно завершены, необходимо создать соответствующие условия, в частности, титрование необходимо проводить относительно медленно, при определенном значении рН среды и температуры. Повышение концентрации ионов водорода и температуры способствует более быстрому течению этой реакции. Скорость реакции повышается в процессе титрования также за счет каталитического действия образующихся ионов Mn2+ (явление автокатализа).
Кроме того, подкисление титруемого раствора в перманганатометрии необходимо проводить только серной кислотой. Хлороводородную кислоту применять нельзя, так как хлорид-ионы окисляются до Cl2, который частично улетучивается (следовательно, невозможно учесть его эквивалентное окислительное действие на восстановители), что приводит к повышенному расходу KMnO4 при титровании:
10Cl– + 2MnO4– + 16H+ = 5Cl2 + 2Mn2+ + 8H2O.
Особенно этот процесс ускоряется в присутствии ионов Fe2+ за счет протекания сопряженной (индуцированной) реакции.
Азотная кислота, являющаяся окислителем, для подкисления в методах редоксиметрии не применяется.
При титровании раствором KMnO4 следует пользоваться бюреткой со стеклянным краном, так как резина окисляется.
Титрованный раствор KMnO4 по точной навеске кристаллического препарата приготовить невозможно, так как в нем всегда содержится некоторое количество MnO2 и другие продукты разложения. Раствор перманганата калия неустойчив из-за реакции с водой, катализируемой оксидом марганца (IV) на свету:
4MnO4– + 2H2O = 4MnO2↓ + 3O2 + 4OH–.
Поэтому раствор перманганата калия следует готовить используя чистую воду, так как органические примеси в воде могут реагировать с MnO4– и давать MnO2. Перед установлением точной концентрации раствор KMnO4 необходимо выдержать в темной склянке в течение 7-10 дней для окончания протекания всех процессов (для ускорения процесса раствор KMnO4 иногда кипятят). Затем осадок MnO2 следует удалить путем фильтрования через стеклянный фильтр. Приготовленный таким образом рас-

65
твор перманганата калия с нормальной концентрацией не ниже 0,05 моль/дм3 (fэкв = 1/5) не изменяет титр продолжительное время.
Стандартизацию раствора перманганата калия проводят по оксалату натрия, дигидрату щавелевой кислоты H2C2O4 · 2H2O, K4[Fe(CN)6], As2O3, соли Мора (NH4)2Fe(SO4)2 · 6H2O, металлическому железу.
Реакция окисления щавелевой кислоты перманганатом калия протекает по уравнению
5H2C2O4 + 2KMnO4 + 3H2SO4 = K2SO4 + 2MnSO4 + 10CO2 + 8H2O, 2 MnO4– + 8H+ + 5ē = Mn2+ + 4H2O
5 C2O42- – 2ē = 2CO2
5C2O42- + 2MnO4– + 16H+ = 10CO2 + 2Mn2+ + 8H2O.
Однако это уравнение выражает только суммарный результат реакции. Реакция между перманганат- и оксалат-ионами протекает очень медленно, поэтому титрование проводят при нагревании раствора щавелевой кислоты до 70-80 0С. Нагревание раствора выше этой температуры может привести к частичному разложению щавелевой кислоты:
H2C2O4 = H2O + CO2 + CO.
Примеры определений перманганатометрическим методом
Определение Fe (II) в гептагидрате сульфата железа (II) FeSO4 · 7H2O и соли Мора (NH4)2Fe(SO4)2 · 6H2O. При титровании перманганатом калия железо (II) окисляется до железа (III):
|
Fe2+ – ē = Fe3+, |
E0 = 0,77 В, |
5Fe2+ + MnO4– + 8H+ = 5Fe3+ + Mn2+ + 4H2O.
Фактор эквивалентности для железа равен единице. Молярная масса соли Мора составляет 392,14 г/моль, молярная масса эквивалента равна молярной массе. Большая эквивалентная масса соли Мора позволяет проводить опреде-
ление методом отдельных навесок.
Рассчитывают массу навески соли Мора для единичного определения, с учетом, что с(1/5KMnO4) = 0,05 моль/дм3 и на титрование должно пойти примерно 20 см3 титранта.
|
m(солиМора) |
392,14 |
0,05 |
20 |
0,3921 0,4 г . |
|
1000 |
||||
Отвешивают 0,4 г соли Мора на аптечных весах, помещают навеску в бюкс, взвешивают на аналитических весах, переносят соль в коническую колбу для титрования и взвешивают пустой бюкс. По разнице масс бюкса с навеской и без навески находят точную массу соли Мора. В колбу добавляют примерно 20 см3 дистиллированной воды, перемешивают до растворения соли. Затем добавляют 20 см3 2н. раствора H2SO4 и титруют 0,05 н. раствором KMnO4 до появления бледно-малинового окрашивания, устойчивого в течение 30 с. В конце титрования новую каплю титранта прибавляют только по-

66
сле того, как исчезнет окраска от последней капли. Рассчитывают массовую долю (%) Fe в соли. M(Fe) = 55,847 г/моль.
|
(Fe) |
c[(1/ 5)KMnO4 |
] M (Fe) V (KMnO4 ) 100 |
. |
|
|
1000 |
m(соли) |
|||
Проводят 2-3 таких определения и рассчитывают среднее значение массовой доли (в %).
Это же определение можно провести методом пипетирования, тогда соответствующую массу навески соли помещают в мерную колбу, растворяют в воде, добавляют раствор H2SO4 и затем объем доводят водой до метки. Аликвоту уже подкисленного раствора соли железа титруют перманганатом калия.
Определение нитритов. Нитрит-ионы окисляются перманганатом калия до нитрат-ионов.
5NaNO2 + 2KMnO4 + 3H2SO4 = 5NaNO3 + 2MnSO4 + K2SO4 + 3H2O,
5 NO2– + H2O – 2ē = NO3– + 2H+
2 MnO4– + 8H+ + 5ē = Mn2+ + 4H2O
5NO2– + 2MnO4– + 6H+ = 5NO3– + 2Mn2+ + 3H2O.
Особенностью рассматриваемого определения является то обстоятельство, что нитриты легко разлагаются кислотами с образованием оксидов азота:
2NO2– + 2H+ = 2HNO2 = NO↑ + NO2↑+ H2O.
Поэтому, чтобы избежать потерь, приходится применять обратный порядок титрования (реверсивное титрование). Подкисленный раствор перманганата калия титруют нейтральным раствором нитрита. При этом нитрит, попадая в раствор KMnO4, практически мгновенно окисляется до нитрата, и оксида азота не образуется. Титрование проводят до обесцвечивания раствора.
Можно также вводить раствор нитрита в подкисленный титрованный раствор KMnO4, взятый в избытке. Остаток KMnO4 затем определяют иодометрически по обратному титрованию.
Определение пероксида водорода. В реакции с KMnO4 пероксид водоро-
да проявляет свойства восстановителя и окисляется до О2:
5H2O2 + 2KMnO4 + 3H2SO4 = 5O2 + 2MnSO4 + K2SO4 + 8H2O, 5 H2O2 – 2ē = O2 + 2H+
2 MnO4– + 8H+ + 5ē = Mn2+ + 4H2O.
Торговый препарат пероксида водорода (пергидроль) содержит около 30% H2O2. Первоначально необходимо установить плотность исследуемого раствора, рассчитать массу навески и соответствующий объем, необходимый для приготовления в мерной колбе приблизительно 0,05н. раствора. Рассчитанный объем пергидроля отмеряют пипеткой с резиновой грушей и переносят в предварительно взвешенный бюкс, определяют массу бюкса с навеской, взвешивая его на тех же весах. Содержимое бюкса количественно (смывая водой несколько раз) переносят в мерную колбу, добавляют воды до полови-

67
ны ее объема, перемешивают, добавляют объем водой до метки и вновь перемешивают.
В колбу для титрования переносят 10,00 см3 разбавленного раствора, добавляют 15 см3 2н. раствора H2SO4, 15-20 см3 воды, перемешивают и титруют 0,05н. раствором KMnO4 до появления бледно-малиновой окраски раствора.
Титрование проводят до трех сходящихся результатов и рассчитывают массовую долю (%) H2O2 в образце:
|
__ |
||||||
|
(H |
2O2 ) |
c[(1/ 5)KMnO4 |
] M[(1/ 2)H 2O2 |
] V (KMnO4 ) Vобщ |
100 |
. |
|
1000 |
Va |
m(H 2O2 ) |
||||
Определение содержания кальция в растворе. Перманганатометриче-
ское определение кальция возможно только косвенными методами, так как катион Са2+ не может быть восстановителем, следовательно, не взаимодействует с KMnO4. Определение Са2+ проводят заместительным титрованием.
По заместительному титрованию из анализируемого раствора ионы кальция осаждают действием щавелевой кислоты:
Са2+ + С2O42- = CaC2O4↓.
Осадок оксалата кальция отфильтровывают, промывают и обрабатывают горячим раствором серной кислоты. При этом в раствор переходит эквивалентное кальцию количество щавелевой кислоты:
CaC2O4 + H2SO4 = CaSO4 + H2C2O4.
Образовавшуюся щавелевую кислоту титруют перманганатом калия.
5H2C2O4 + 2KMnO4 + 3H2SO4 = 10CO2 + 2MnSO4 + K2SO4 + 8H2O.
Фактор эквивалентности для оксалат-иона равен 1/2, следовательно, и для Са2+, взаимодействующего с оксалатом-ионом в соотношении 1:1, соответственно равен 1/2.
Метод перманганатометрии является одним из распространенных методов редоксиметрии. Его широко используют для анализа лекарственных и биологических препаратов, воды и пищевых продуктов. Так, этим методом определяют сахара в хлебобулочных изделиях, примеси KJ в поваренной соли. По количественному содержанию кальция рассчитывают количество молока в напитках (кофе, какао). Перманганатометрию используют для определения общей окисляемости воды и почвы.
4.3. Дихроматометрия
Дихроматометрическое титрование основано на взаимодействии определяемых веществ с дихроматом калия. Основной реакцией метода является реакция окисления восстановителей дихроматом калия в кислой среде.
|
68 |
|
|
Cr2O72- + 14H+ + 6ē = 2Cr3+ + 7H2O, |
E0 = 1,33 В. |
Как показывает величина стандартного окислительно-восстановительного потенциала, дихромат калия является менее сильным окислителем, чем
KMnO4, но тем не менее дихроматометрию успешно применяют для определения многих восстановителей: Fe2+, Mn2+, Sb3+, Sn2+, SO32-, AsO33-, J–,
[Fe(CN)6]4-, спиртов, глицерина, аскорбиновой кислоты и др. Например:
Cr2O72- + 6[Fe(CN)6]4- + 14H+ = 2Cr3+ + 6[Fe(CN)6]3- + 7H2O,
Cr2O72- + CH3OH + 8H+ = 2Cr3+ + CO2 + 6H2O, 2Cr2O72- + 3C2H5OH + 16H+ = 4Cr3+ + 3CH3COOH + 11H2O.
Достоинством дихраматометрического метода является то, что титрованный раствор можно приготовить по точной навеске, поскольку K2Cr2O7 удовлетворяет всем требования первичного стандарта. Раствор K2Cr2O7 очень устойчив. Дихромат калия не окисляет (без нагревания) хлоридионы, что позволяет проводить титрование в соляно-кислой среде.
Для фиксирования точки эквивалентности чаще всего используют редокс-индикаторы: дифениламин и его производные (дифениламиносульфоновую, фенилантраниловую кислоты).
Механизм реакций с участием ионов Cr2O72- сложен. В случае медленного протекания реакции прибегают к обратному титрованию, при нагревании избыток K2Cr2O7 оттитровывают раствором соли железа (II). По обратному титрованию определяют и окислители, которые предварительно восстанавливают избытком титранта (Fe2+), остаток которого титруют раствором K2Cr2O7:
3Fe2+ + NO3– + H+ = 3Fe3+ + NO↑ + 2H2O,
6Fe2+ + Cr2O72- + 14H+ = 6Fe3+ + 2Cr2- + 7H2O.
Иногда используют осадительный вариант метода (заместительное титрование) для анализа солей Ag+, Ba2+, Pb2+, образующих нерастворимые осадки хроматов.
4.4.Иодометрия
Воснове иодометрического титрования лежат реакции восстановления свободного иода до иодид-ионов и окисления иодид-ионов в свободный иод (реакция обратима):
|
J2 + 2ē 2J –, |
E0 = 0,62 В. |
Иод плохо растворим в воде, но в присутствии иодид-иона образуется комплексный ион J3–, поэтому при титровании протекает реакция:
|
J3 |
– + 2ē 3J –, E0 = 0,54 В. |
Хорошая обратимость реакции и невысокий стандартный потенциал E0(J3–/3J–) позволяют определять иодометрически восстановители, стан-
69
дартный потенциал, которых меньше 0,54 В, и окислители, потенциал которых больше потенциала пары иода.
При прямом определении восстановителей рабочим стандартным раствором случит раствор иода, который готовят растворением очищенного путем возгонки J2 в растворе KJ. Хранят раствор в темной склянке (с плотно пришлифованной стеклянной пробкой) во избежание окисления иодида (окисление иодид-ионов кислородом воздуха ускоряется на свету) и улетучивания образовавшегося иода. Концентрацию раствора иода можно проверить по ПСВ, например, As2O3.
Метод титрования раствором иода иногда называют иодиметрией. Его используют для определения Sn (II), As (III), Sb (III), S2-, SO32-, S2O32-, орга-
нических соединений, например, аскорбиновой кислоты, сахаров, спиртов, альдегидов. Определение некоторых восстановителей по ряду причин проводят по обратному титрованию. Вторым титрантом служит раствор тиосульфата натрия, который окисляется до тетратионата натрия. Например, определение сульфитов основано на реакциях:
Na2SO32- + J2 + H2O = Na2SO4 + 2HJ,
J2 + 2Na2S2O3 = 2NaJ + Na2S4O6.
Прямая реакция восстановления иода идет быстро, но обратная реакция окисления иодида протекает медленнее. Поэтому использовать раствор иодида для определения окислителей путем прямого титрования невозможно. К тому же раствор KJ неустойчив, поскольку иодид-ион окисляется кислородом воздуха. В этом случае используют заместительное титрование – добавляют к окислителю избыток иодида, а выделившийся в эквивалентном количестве иод оттитровывают стандартным раствором тиосульфата натрия. Этот метод называют иодометрия (следует отметить, что терминология разделения на иодиметрию и иодометрию соблюдается не строго и обе группы методов часто называют иодометрическими). Иодометрическим методом определяют окислители: KMnO4, K2Cr2O7, KClO3, NaNO2, H2O2, Cl2, Br2, соли меди (II), железа (III) и др.
Кислородосодержащие окислители взаимодействуют с избытком иодида калия в кислой среде, например перманганат-ион:
2MnO4– + 10J– + 16H+ = 2Mn2+ + 5J2 + 8H2O, J2 + 2S2O32- = 2J– + S4O62-.
Титровать окислители (кроме J2) непосредственно тиосульфатом натрия нельзя, так как во многих случаях реакция нестехиометрична и невозможно использовать специфический индикатор метода – крахмал.
Раствор тиосульфата натрия является вторичным стандартным раствором, который готовят из пентагидрата тиосульфата натрия Na2S2O3 · 5H2O. Раствор неустойчив. В нем возможно окисление кислородом воздуха S2O32- до SO42- и S, а также образование HSO3– (под влиянием углекислоты). Таким образом, свежеприготовленный раствор Na2S2O3 первое время (7-10

70
дней) медленно меняет свой титр. Добавление Na2CO3 и предохранение от CO2 с помощью хлоркальциевой трубки стабилизирует раствор. Рекомендуется также добавлять немного фенола или хлорамина для уничтожения серных бактерий, способствующих разложению реагента.
Для стандартизации раствора тиосульфата натрия обычно используется дихромат калия:
K2Cr2O7 + 6KJ + 7H2SO4 = Cr2(SO4)3 + 3J2 + 4K2SO4 + 7H2O, 1 Cr2O72- + 14H+ + 6ē = 2Cr3+ + 7H2O
3 2J– – 2ē = J2.
Выделившийся через несколько минут иод титруют тиосульфатом натрия
2Na2S2O3 + J2 = 2NaJ + Na2S4O6,
1 2S2O3– – 2ē = S4O62-
1 J2 + 2ē = 2J–.
Из ионного уравнения реакции видно, что два тиосульфата-иона S2O32- превращаются в один тетратионат-ион S4O62-, отдавая молекуле иода два электрона. Следовательно, фактор эквивалентности тиосульфата натрия равен (2/2) единице.
Стандартизованный 0,05-0,02М раствор тиосульфата натрия хранят в склянках из темного стекла, при рассеянном свете или в темноте. Титр раствора проверяют еженедельно. Если раствор помутнел, его выливают, так как выпадение серы свидетельствует о существенном разложении тиосульфата натрия.
Фиксирование точки эквивалентности в иодометрии возможно без добавления индикатора по обесцвечиваю желтой окраски иода при его титровании тиосульфатом или по появлению желтой окраски от одной кали иода при прямом титровании восстановителей. Однако этот способ недостаточно чувствителен. При титровании разбавленных или окрашенных растворов в качестве индикатора используют крахмал. Крахмал не растворяется в воде. Но при нагревании его взвеси до кипения получается коллоидный раствор, который и используется в иодометрии. Чувствительность крахмала к иоду значительно повышается в присутствии иодид-ионов (до 10-5М). В коллоидном состоянии крахмал образует с полигалид-ионом [J3]– адсорбционное соединение синего цвета.
При работе с крахмалом в качестве индикатора следует учитывать некоторые особенности. К раствору, содержащему иод, крахмал добавляют в конце титрования, когда раствор приобретет слабую соломенно-желтую окраску. Объясняется это не только тем, что крупные сгустки крахмала медленно отдают адсорбированный иод и плохо обесцвечиваются тиосульфатом. Помимо этого, крахмал обладает свойством частично восстанавливать некоторые окислители. Поэтому вблизи точки конца титрования необходимо добавлять раствор тиосульфата натрия по одной капле, после каждой капли тщательно перемешивать и ждать обесцвечивания раствора
71
в течение 3-5 с. Если не соблюдать эти условия, раствор очень легко перетитровать. При прямом титровании восстановителей, например хлорида олова (II), раствор крахмала приливают с самого начала титрования и титруют до появления синего окрашивания.
В качестве индикатора используют свежеприготовленный 0,2%-0,5% раствор крахмала. Для повышения устойчивости (раствор быстро разлагается микроорганизмами) раствор стерилизуют в течение 2-3 часов на водяной бане или к раствору добавляют антисептики (ZnCl2, HgJ2, салициловую кислоту). Обычно на 50 см3 титруемого раствора добавляют 2-3 см3 индикатора.
Иодометрическое титрование необходимо проводить при определенных условиях. Прямое титрование иодом обычно проводят в нейтральной или кислой среде. При рН > 11 иод окисляется до гипоиодита JO–:
J2 + 2OH– = JO– + J– + H2O.
Гипоиодит-ион является более сильным окислителем, чем J2, дает побочные реакции, например окисляет тиосульфат-ион до сульфата:
S2O32- + 4JO– + 2OH = 4J– + 2SO42- + H2O.
Поэтому титровать в щелочной среде (рН > 8) не рекомендуется.
В большинстве методик окисление органических веществ проводят в щелочной среде, то есть гипоиодитом, но после окончания реакции раствор подкисляют и избыток вновь выделившегося иода оттитровывают тиосульфатом. Титрование в сильнокислой среде может привести к возникновению погрешности, связанной с частичным окислением J– кислородом воздуха. Кроме того, титрование окислителей проводят при избытке иодида калия, который необходим для растворения J2. Так как скорость взаимодействия окислителей с KJ невелика, то дают время (10-15 минут) для завершения реакции. При этом реакционную смесь в закрытой колбе оставляют в темном месте. Титрование проводят при комнатной температуре, так как при нагревании J2 улетучивается и, кроме того, понижается чувствительность иод-крахмальной реакции.
Возможности иодометрического метода распространяются не только на реакции окисления-восстановления, но и на реакции присоединения, замещения и др. Так, реакции присоединения широко применяются в аналитической химии органических соединений для количественного определения двойных связей в ненасыщенных соединениях:
> C = C < + J2 = > CJ ─ CJ < .
Степень ненасыщенности определяется иодным числом, которое служит одной из характеристик, например, жидких жиров.
Иод способен замещать атомы водорода в ароматических соединениях, например, фенолах, ароматических диаминах и др.
Иодометрические определения отличаются большим разнообразием и этим объясняется широкое применение метода в практике различных ла-
![]()
72
бораторий. Так, метод иодометрии применяют для определения альдегидов и кетонов в объектах пищевой промышленности, бисульфитсвязывающих соединений, иодного числа, используемого главным образом для определения липидов, жирных кислот и при определении содержания реагирующих с иодом примесей в ароматических углеводородах; редуцирующих веществ в различных пищевых продуктах, сернистой и аскорбиновой кислоты во фруктов-ягодных полуфабрикатах и напитках.
Определение массовой доли (%) аскорбиновой кислоты в растворе
Метод основан на окислении аскорбиновой кислоты (АК) иодом в кислой среде до дегидроаскорбиновой кислоты (ДГАК):
C6H8O6 + J2 = C6H6O6 + 2HJ,
C6H8O6 – 2ē = C6H6O6 + 2H+, J2 + 2ē = 2J– .
Стандартный потенциал ЕДГАК/АК при рН 1 составляет 0,33 В. Фактор эквивалентности аскорбиновой кислоты равен 1/2. М(АК)= 176,0 г/моль.
Обычно используют обратное титрование. Не участвующий в реакции остаток иода оттитровывают тиосульфатом натрия с индикатором – крахмалом:
J2 + 2Na2S2O3 = 2NaJ + Na2S4O6.
Для определения приблизительной концентрации анализируемого раствора определяют его плотность по ареометру. Допустим, плотность раствора ρ = 1,035 г/см3, ей соответствует массовая доля – 8,5 %. Нормальную концентрацию раствора находят по формуле 2.12 б:
|
с(1/ 2) А.К. |
8,5 10 1,035 |
0,999 1 моль / дм3 . |
|
88,0 |
||
Так как при титровании используют 0,05н. раствор иода, необходимо разбавить исследуемый раствор до концентрации титранта. Находят объем концентрированного раствора Vконц, необходимый для приготовления, допустим, 100,0 см3 разбавленного (~ 0,05н.) раствора аскорбиновой кислоты:
|
V |
0,05 100,0 |
5,0 см3 . |
||
|
конц |
||||
|
1 |
||||
Рассчитанный объем с помощью измерительной пипетки на 5,00 см3 переносят в мерную колбу вместимостью 100,0 см3, добавляют объем водой до метки и вновь перемешивают. Далее определение проводят по следующей методике. В коническую колбу для титрования переносят пипеткой 10,00 см3 разбавленного раствора аскорбиновой кислоты, добавляют с помощью цилиндра 4 см3 6 М раствора H2SO4, вводят из бюретки 20,00 см3 0,05н. раствора иода. Колбу закрывают пробкой, оставляют в темном месте на 3-5 минут и затем титруют избыток иода 0,05 М раствором Na2S2O3. Сначала титруют без индикатора. Когда окраска из темно-бурой превра-

73
тится в соломенно-желтую, прибавляют 1-2 см3 0,5 % раствора крахмала и (не доливая объема бюретки) продолжают медленно титровать, хорошо перемешивая раствор, до исчезновения синей окраски.
Титрование повторяют до трех сходящихся результатов. Находят средний объем тиосульфата натрия и рассчитывают массовую долю (в %) аскорбиновой кислоты в исходном образце:
|
ω(A.K.) |
[c(1/2J |
2 ) V(J |
2 ) c(Na 2 S2 O3 |
) V(Na2 S2O3 )] M(1/2A.K.) Vобщ |
100 |
. |
|
1000 |
Va |
ρ Vконц |
||||
Контрольные вопросы
1.Каким требованиям должны отвечать окислительно-восстановительные реакции, лежащие в основе количественных определений?
2.Какая реакция лежит в основе перманганатометрического титрования?
3.Почему перманганатометрическое титрование проводят, в основном, в кислой среде? Какая кислота при этом используется?
4.Каким образом определяют точку эквивалентности в методе перманганатометрии?
5.Как готовится титрованный раствор перманганата калия? Какие ПСВ используются для его стандартизации?
6.Какое вещество является катализатором при перманганатометрических определениях?
7.Уравняйте реакцию и определите фактор эквивалентности для окислителя и восстановителя:
H2S + KMnO4 + H2SO4 = S + MnSO4 + K2SO4 + H2O.
8.Какая реакция лежит в основе дихроматометрии?
9.В чем заключается сущность иодометрии?
10.Почему в перманганатометрии и дихроматометрии используются лишь окисленные, но не восстановленные формы соответствующих редокспар? Почему в отличие от этого в идометрии используются обе формы пары?
11.Как проводят определение окислителей и восстановителей иодометрическим методом?
12.Какие условия необходимо соблюдать при иодометрических определениях?
13.Каковы особенности приготовления титрованных растворов иода и тиосульфата натрия?
14.Почему при иодометрическом определении окислителей используется большой избыток KJ?
15.В чем заключаются особенности использования крахмала в качестве индикатора?
74
ГЛАВА 5.2. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
5.1. Осадительное титрование
Методы осадительного титрования основаны на реакциях осаждения. Не все реакции осаждения пригодны для титриметрических определений, а только те из них, которые удовлетворяют ряду требований:
1)реакция должна протекать стехиометрично (обычно используют реакции, где ионы взаимодействуют в соотношении 1:1);
2)осадок должен быть практически не растворим;
3)осадок должен выпадать быстро;
4)явления соосаждения не должны заметно искажать результаты титрования;
5)должна иметься возможность фиксировать точку эквивалентности. Удовлетворительны с этой точки зрения реакции осаждения галогени-
дов и тиоцианата нитратом серебра или нитратом ртути (I). Методы аргентометриии применяют для анализа солей галогеноводородных кис- лот-хлоридов, бромидов и иодидов щелочных и щелочноземельных металлов и органических оснований. Соли серебра определяют путем осаждения их тиоцианатом аммония или калия (тиоцианатометрия). Реже применяют меркурометрическое определение галогенидов.
5.1.1. Аргентометрическое титрование
Основными стандартными растворами аргентометрии являются нитрат серебра и тиоцианат аммония. Хранят раствор AgNO3 в склянках из темного стекла, так как на свету соли серебра неустойчивы. Стандартизацию раствора AgNO3 обычно проводят по NaCl. Точную концентрацию NH4NCS (или KNCS) устанавливают по вторичному стандартному раство-
ру AgNO3.
Конечная точка титрования в аргентометрии может быть установлена различными способами, вошедшими в химическую литературу под именами ученых, которые их разработали: методы Гей-Люссака, Мора, Фольгарда, Фаянса.
Так, по методу Гей-Люссака индикатор не используют, титрование проводят до просветления, когда новая порция осадителя не вызывает помутнения раствора. Метод достаточно точный, но требует много времени, и в настоящее время практически не применяется.
Метод Мора. В качестве индикатора в методе Мора используют хромат калия K2CrO4. Хромат-ионы образуют с ионами серебра кирпичнокрасный осадок Ag2CrO4, более растворимый, чем галогениды серебра
75
[P(Ag2CrO4) = 6,5 · 10-5 моль/дм3, P(AgCl) = 1,3 · 10-5 моль/дм3, P(AgBr) =
7,2 · 10-7 моль/дм3].
При титровании хромат серебра не образуется, пока не будет оттитрован галогенид-ион:
Cl– + Ag+ = AgCl↓,
CrO42- + 2Ag+ = Ag2CrO4↓.
Важно правильно выбрать концентрацию хромат-ионов. Если она будет очень мала, потребуется большой избыток серебра для образования заметного визуально осадка. При слишком большой концентрации CrO42- образование осадка начинается раньше, чем оттитруется галогенид-ион. На практике создают концентрацию CrO42-, равную 0,01 – 0,05 моль/дм3.
Титрование с хроматом в качестве индикатора проводится в нейтральной или слабощелочной среде, когда рН раствора больше 6,5; но менее 10,5. В более кислой среде происходит протонирование хромата и чувствительность индикатора падает:
CrO42- + H+ = HСrO4– и 2HCrO4– = Cr2O72- + H2O.
В щелочной среде может выпадать осадок оксида серебра (раньше, чем хромата серебра):
Ag+ + OH– = AgOH↓, 2AgOH = Ag2O↓ + H2O.
В аммиачной среде хлорид и хромат серебра растворяются с образованием комплексного аммиаката серебра.
Метод Мора применим для определения хлоридов и бромидов. Иодиды и тиоционаты не определяются, так как вследствие их адсорбции на поверхности осадка установление точки эквивалентности становится затруднительным. Определению по методу Мора мешают ионы, образующие осадки с хроматом или ионом серебра, а также окрашенные ионы.
Метод Мора широко применяется при анализе пищевых продуктов. Этим методом определяют содержание поваренной соли в отдельных продуктах или рассолах.
Метод Фольгарда. Метод основан на титровании ионов серебра раствором тиоционата аммония в присутствии ионов Fe (III) в качестве индикатора:
Ag+ + NCS– = AgNCS↓,
Fe3+ + NCS– = [Fe(NCS)]2+.
После оттитрования ионов серебра, лишняя капля титранта NH4NCS образует с ионами Fe3+ тиоцианатный комплекс и раствор приобретает оранжевую окраску. В качестве индикатора используют железоаммонийные квасцы (NH4)2Fe(SO4)2 · 12H2O или другие соли Fe (III). Обычно создают концентрацию Fe3+ около 0,01 моль/дм3. Титрование проводят в кислой среде (для подавления гидролиза соли железа).
Для определения анионов этим методом (Cl–, Br–, J–) используют обратное титрование. К раствору титруемого иона добавляют избыток стан-
76
дартного раствора нитрата серебра, после образования осадка оттитровывают избыток AgNO3 стандартным раствором NH4NCS в присутствии соли железа (III). При образовании осадков менее растворимых, чем AgNCS (ПР = 1,1 · 10-12), возможно непосредственное титрование избытка Ag+ над осадком (определение J–, Br–), но при определении Cl– более растворимый AgCl (ПР = 1,78 · 10-10) может частично перейти в AgNCS, поэтому необходимо отделить его (фильтрованием или добавлением тяжелого органического растворителя, например нитробензола, покрывающего осадок и предохраняющего его от контакта с раствором):
Br– + Ag+ = AgBr↓,
Ag+ + NCS– = AgNCS↓,
NCS– + Fe3+ = [Fe(NCS)]2+.
Существенным достоинством метода Фольгарда является возможность определения галогенидов в кислой среде.
Метод Фаянса. В методе Фаянса используют адсорбционные индикаторы, которые при адсорбции на осадке изменяют цвет. Установлено, что в первую очередь на осадке адсорбируются ионы, одноименные с осадком и находящиеся в избытке. Например, при титровании хлорида натрия нитратом серебра на осадке AgCl до точки эквивалентности будут адсорбироваться преимущественно хлорид-ионы (AgCl/Cl–) и для нейтрализации отрицательного заряда к частицам осадка будут притягиваться ионы Na+ (AgCl/Cl– · Na+). После точки эквивалентности адсорбироваться на осадке будут избыточные ионы Ag+ и для нейтрализации уже положительного заряда осадка из раствора будут притягиваться отрицательно заряженные ионы, в том числе ионы индикатора. Анионы некоторых красителей, адсорбируясь, меняют свой цвет. Например, флуоресцеин – слабая органическая кислота желто-зеленого цвета, диссоциирует с образованием аниона (Фл–), который, адсорбируясь на положительно заряженном осадке (AgCl/Ag+ · Фл–), изменяет свой цвет на розовый. Титрование с флуоресцеином происходит при рН 7-10. При титровании в более кислой среде следует использовать красители с более сильными кислотными свойствами, например тетрабромфлуоресцеин (эозин).
Необходимым условием применения адсорбционного индикатора является его способность служить противоионом, но не вытеснять первичноадсорбированный ион осадка. Однозарядные анионы замещают друг друга на поверхности галогенида серебра в порядке, коррелирующим с поляризуемостью анионов. При рН 7 порядок замещения таков: J–, CN– >NCS– > Br– > анион эозина > Cl– > анион флуоресцеина. Таким образом, при рН 7 анион флуоресцеина будет служить индикатором, если он стоит в этом ряду правее титруемого иона.
77
5.1.2. Меркурометрическое титрование
Метод меркурометрии основан на осаждении галогенид-ионов ионами ртути (I):
2Cl– + Hg22+ = Hg2Cl2↓.
Титрантом метода является раствор нитрата ртути (I), который готовят из реагента Hg2(NO3)2 · 2H2O и стандартизуют по NaCl или NaBr.
В качестве индикаторов в меркурометрии применяют тиоционатные комплексы железа (III) и дифенилкарбазон. Приминение первого индикатора основано на обесцвечивании растворов тиоцианатных комплексов железа (III) под действием иона Hg22+, образующего осадок Hg2(NCS)2:
Cl– + Hg22+ = Hg2Cl2↓, 2[Fe(NCS)]2+ + Hg22+ = Hg2(NCS)2↓ + 2Fe3+.
Обесцвечивание происходит вблизи точки эквивалентности. Объем стандартного раствора Hg2(NO3)2, израсходованный на реакцию с индикатором, устанавливают проведением холостого опыта.
Дифенилкарбазон, возможно, являясь адсорбционным индикатором, в точке экивалентности окрашивает осадок Hg2Cl2 в ярко-голубой или синефиолетовый цвет. Титрование с дифенилкарбазоном проводят в сильнокислой среде, что является достоинством метода.
Меркурометрический метод применяют для определения хлоридов и бромидов. Титрование может проводиться в разбавленных (0,01 М) растворах.
Недостатками методов аргентометрии и меркурометрии является высокая стоимость нитрата серебра и токсичность солей ртути.
5.2. Комплексиметрическое титрование
5.2.1. Разновидности комплексиметрии
Комплексиметрическое титрование основано на использовании реакций образования комплексных соединений при титровании ионов металлов раствором комплексообразующего реагента. Среди реакций с участием неорганических лигандов в титриметрии применяют реакции образования галогенидов ртути (II), фторидов алюминия, тория и цианидов некоторых тяжелых металлов (никель, кобальт, цинк). На образовании этих комплек-
сов основаны методы меркуриметрии, фторидометрии и цианидомет-
рии.
Недостатком использования в комплексиметрии монодентатных неорганических и органических лигандов является многоступечатость образования комплексов, в результате чего на кривой титрования часто отсут-

78
ствует скачок. Многие же полидентатные лиганды реагируют с ионами металлов в простом стехиометрическом соотношении 1:1.
Титрование с использованием полидентатных органических реагентов называют комплексонометрией. В качестве таких аналитических органических реагентов используют комплексоны – соединения, являющиеся производными аминополикарбоновых кислот. К ним относятся иминодиуксусная кислота, нитрилтриуксусная кислота (комплексон I), этилендиаминатетрауксусная кислота или ее динатриевая соль и многие другие.
Наиболее часто применяют этилендиаминотетрауксусную кислоту
(комплексон II) – ЭДТУ:
|
HOOCH2C |
CH2COOH |
|
N – CH2 – CH2 – N |
|
|
HOOCH2C |
CH2COOH |
Анион ее обычно обозначают символом Y (с соответствующим зарядом и степенью протонирования). Так как ЭДТУ плохо растворима в воде, на практике используют хорошо растворимую ее соль Na2H2Y·2H2O (комплексон III) – ЭДТА.
5.2.2. Комплексонометрия
Титрованный раствор ЭДТА готовят обычно из продажного препарата, проверку концентрации осуществляют по стандартным растворам цинка (II) или висмута (III), приготовленным растворением соответствующих металлов в соляной кислоте.
Как гексадентатный лиганд, ЭДТА образует с ионами многих металлов (в том числе и щелочноземельных, которые трудно перевести в комплекс) растворимые в воде прочные внутрикомплексные соединения, относящиеся к группе хелатов. Комплексы образуются путем замещения ионов водорода карбоксильных групп в молекуле ЭДТА ионом металла и одновременного взаимодействия с атомами азота аминогрупп (за счет донорноакцепторной связи). Например, ион металла М2+ образует при комплексообразовании три устойчивых цикла:
Ион М3+ замыкает еще одно кольцо с СООН-группой. Такой комплекс еще более прочен:
79
|
O = C – O |
O – C = O |
|||||
|
M2+ |
||||||
|
H2C |
CH2 |
|||||
|
N |
N |
|||||
|
HOOCH2C |
H2C |
CH2 |
CH2COOH |
|||
Схематично взаимодействие комплексона III с ионами металлов можно выразить следующими уравнениями:
M2+ + H2Y2- = MY2- + 2H+,
M3+ + H2Y2- = MY– + 2H+,
M4+ + H2Y2- = MY + 2H+.
Из приведенных уравнений видно, что одна молекула ЭДТА реагирует с одним катионом металла (независимо от степени его окисления), т.е. стехиометрия реакции 1:1. Так как в результате всех реакций выделяется два иона водорода, фактор эквивалентности для ЭДТА и ионов металлов, равен 1/2. Учитывая стехиометрию реакции, чаще для расчетов в комплексонометрии используют молярные концентрации.
Помимо основной реакции комплексообразования, в системе М-ЭДТА- Н2О протекают конкурентные реакции протонирования ЭДТА и образования гидроксокомплексов металлов. Комплексы ионов металлов с ЭДТА характеризуются константами устойчивости, причем, как правило, чем выше заряд иона, тем прочнее комплекс. На прочность комплексов влияет рН среды. Некоторые комплексы, например, кальция и магния, устойчивы только в щелочной среде. Поскольку в результате реакции рН среды меняется (выделяются ионы Н+), для предотвращения смещения равновесия реакции в обратном направлении титрование проводят в присутствии буферной смеси (чаще аммонийной NH4OH + NH4Cl). Ионы, образующие более прочные комплексы (Zn2+, Pb2+), можно титровать в умереннокислой среде (поддерживая постоянство рН кислыми буферами, например ацетатным), а трех-и четырехзарядные ионы – даже в сильно кислой среде.
Для фиксирования точки эквивалентности в комплексонометрии применяют металлоиндикаторы – органические соединения, которые с ионами металлов образуют окрашенные комплексы. Полученные при этом комплексные соединения менее прочны, чем комплексы металлов с комплексоном. Поэтому при титровании, когда все определяемые катионы связаны в прочные комплексы (точка экивалентности), добавление лишней
80
капли титранта приводит к разрушению комплекса индикатора с ионом металла и наблюдается изменение окраски.
Наибольшее распространение приобрели так называемые металлохромные индикаторы, содержащие в своих молекулах хромофорные группы, следовательно, окрашенные и образующие с ионами металлов внутрикомплексные соединения, по цвету отличающиеся от самих индикаторов. К таким индикаторам относятся, например, эрихром черный Т, который с ионами металлов Mg2+, Ca2+, Zn2+ и другими образует комплексы винно-красного цвета; в то время как сам индикатор при рН 8-10 имеет синий цвет, хромоген темно-синий также имеет синий цвет в щелочной среде, а его комплексы с ионами металлов – розового цвета.
Как и в других титриметрических методах, для получения правильных результатов в комплексонометрии необходимо, чтобы рТ индикатора входил в скачок на кривой титрования.
Определение ионов металлов проводят прямым или обратным титрованием.
Прямое титрование проводится при достаточно высокой скорости образования комплексоната металла, отсутствии побочных реакций, доста-
точно контрастном переходе окраски индикатора. Так определяют большинство ионов металлов, например, Zn2+, Mg2+, Ca2+, Ba2+, Cd2+ при рН 8-
10 с индикатором эриохромом черным Т, Fe3+ при рН 1-2 с ксиленоловым оранжевым и др.
Схематично прямое титрование с металлохромным индикатором можно представить следующим образом:
M2+ + Ind = MInd,
M2+ + H2Y2_ = MY2- + 2H+,
MInd + H2Y2- = MY2- + Ind + 2H+.
Обратное титрование, основанное на добавлении избыточного количества ЭДТА и последующем титровании избытка стандартным раствором другого металла, применяют в тех случаях, когда реакции образования комплекса с ЭДТА протекают медленно или нет подходящего металлоиндикатора для фиксирования точки эквивалентности. Например, при определении соли Al3+, хрома (III), ртути (II). Для ускорения реакции иногда реакционную смесь нагревают до кипения (определение Al3+, Cr3+), затем охлаждают и титруют солью магния или цинка:
Al3+ + H2Y2- = AlY– + 2H+,
H2Y2- + Mg2+ = MgY2- + 2H+,
Mg2+ + Ind = MgInd.
Косвенное титрование применяют для определения элементов, не образующих комплексов с ЭДТА. Так при определении анионов их осаждают избытком стандартного раствора какого-либо иона металла, осадок отде-
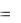
81
ляют и в фильтрате титруют избыток ионов металла, не осадившихся определяемым анионом. Например, при определении SO42- их осаждают избытком BaCl2 (в виде BaSO4), а несвязанные ионы бария оттитровывают комплексоном III. Фосфат-ионы осаждают в виде MgNH4PO4, избыток магния определяют компексонометрически. Оксалат-ионы осаждают в ви-
де CaC2O4.
Комплексонометрическое титрование применяют для определения общей жесткости воды (Ж0). Жесткость воды характеризуется молярной концентрацией эквивалентов кальция и магния (fэкв = 1/2) и выражают в моль/дм3. Содержание этих элементов определяют прямым титрованием пробы воды V(H2O) в аммонийном буфере 0,01 М раствором ЭДТА в присутствии эриохром черного Т, как индикатора, и рассчитывают по формуле
|
Жо |
с(1/ 2Na2 H 2Y ) V (Na2 H 2Y ) 1000 |
. |
|
|
V (H |
2O) |
||
Метод комплексонометрии используется для определения Mg2+ и Ca2+ в различных соках на сахароваренных заводах и предприятиях пищевой промышленности, а также в почве, удобрениях, растительных и животных тканях, молоке, крови и т.д.
Контрольные вопросы
1.Каким условиям должны удовлетворять реакции, используемые в методах осаждения?
2.В каких методах осадительного титрования в качестве индикатора используются реакции образования: а) окрашенных малорастворимых соединений; б) окрашенных комплексных соединений; в) окрашенных адсорбционных соединений?
3.На чем основаны методы Мора, Фольгарда и Фанса?
4.В каких условиях проводят аргентометрическое титрование по Мору?
5.На чем основано меркурометрическое определение хлоридов?
6.На чем основан комплексонометрический метод анализа?
7.Какие соединения называют комплексонами?
8.Какое значение имеет рН при комплексонометрическом титровании?
9.На чем основано действие металлоиндикаторов в комплексонометрии?
10.Какова область применения метода комплексонометрии?
82
Библиографический список
1.Алексеев В.Н. Количественный анализ. – М.: Химия, 1972.
2.Барсукова З.А. Аналитическая химия. – М.: Высшая школа, 1990.
3.Васильев В.П. Аналитическая химия. В 2 ч. Ч.1 Гравиметрический и титриметрический метод анализа. – М.: Высшая школа, 1989.
4.Жванко Ю.Н., Панкратова Г.Р, Мамедова З.И. Аналитическая химия и технохимический контроль в общественном питании. – М.: Высшая школа, 1989.
5.Золотов Ю.А, Дорохова Е.Н., Фадеева В.И. и др. Основы аналитической химии: В 2 кн. Кн. 2. Методы химического анализа. – М.: Высшая школа, 2000.
6.Крешков А.П. Основы аналитической химии: В 3 кн. Кн. 2. Количественный анализ. – М.: Химия, 1976.
7.Лебухов В.И., Окара А.И., Павлюченкова Л.П. Физико-химические свойства и методы контроля качества потребительских товаров. – Хабаровск: РИЦ ХГАЭП, 1999.
8.Павлюченкова Л.П. Аналитическая химия: В 2 ч. Ч. 1. – Качественный анализ. – Хабаровск: РИЦ ХГАЭП, 2000.
9.Петерс Д. Хайес Дж., Хифтье Г. Химическое разделение и измерение. Теория и практика аналитической химии: В 2ч. Ч. 1 Пер. с англ.
– М.: Химия, 1976.
10.Пилипенко А.Т., Пятницкий И.В. Аналитическая химия: В 2 кн.
Кн.1. – М.: Химия, 1990.
11.Полеес М.Э., Душечкина И.Н. Аналитическая химия. – М.: Медици-
на, 1987.
12.Пономарев В.Д. Аналитическая химия: В 2 ч. Ч. 2. Количественный анализ. – М.: Высшая школа, 1982.
13.Попадич И.А., Траубенберг С.Е., Осташенкова Н.В., Лысюк Ф.А. Аналитическая химия. – М .: Химия, 1989.
14.Посыпайко В.И., Козырев Н.А., Логачева Ю.П. Химические методы анализа. – М.: Высшая школа, 1989.
15.Скуг Д., Уэст Д. Основы аналитической химии: В 2 ч. Ч. 2 Пер.с
англ. – М.:Мир, 1979.
16.Толстоусов В.Н., Эфрос С.М. Задачник по количественному анализу.
– Л.: Химия, 1986.
17.Ушакова Н.Н. Курс аналитической химии. – Издательство Московского университета, 1984.
18.Цитович И.К. Курс аналитической химии. – М.: Высшая школа,
1985.
|
83 |
|
|
ОГЛАВЛЕНИЕ |
|
|
Предисловие |
3 |
|
Глава 1. Основные принципы количественного анализа |
4 |
|
1.1. Задачи количественного анализа |
4 |
|
1.2. Принципы и методы количественных определений |
5 |
|
1.3. Правила вычислений в количественном анализе |
6 |
|
1.4. Погрешность количественных определений |
7 |
|
1.5. Аналитические весы и взвешивание |
10 |
|
1.6. Измерительная аналитическая посуда и правило работы с ней |
16 |
|
Глава 2. Титриметрический анализ |
21 |
|
2.1. Сущность титриметрического анализа |
21 |
|
2.2. Классификация методов титриметрического анализа |
22 |
|
2.3. Основные приемы титрования |
23 |
|
2.4. Расчеты в титриметрическом анализе |
25 |
|
2.4.1. Эквивалент |
25 |
|
2.4.2. Способы выражения концентрации веществ в химическом |
|
|
анализе |
28 |
|
2.4.3. Стандартные растворы |
31 |
|
2.4.4. Расчет массы навески исследуемой пробы и вычисление |
|
|
результатов титриметрических определений |
36 |
|
2.5. Кривые титрования |
41 |
|
Глава 3. Кислотно-основное титрование (протолитометрия) |
43 |
|
3.1. Сущность метода |
43 |
|
3.2. Индикаторы кислотно-основного титрования |
45 |
|
3.3. Кривые кислотно-основного титрования и выбор индикатора |
48 |
|
3.4. Титранты метода |
52 |
|
3.4.1. Приготовление и стандартизация растворов кислот и щелочей |
52 |
|
3.5. Примеры определений методом кислотно-основного титрования |
56 |
|
3.5.1. Определение массовой доли (%) NaHCO3 в твердом образце |
56 |
|
3.5.2. Определение массовой доли (%) уксусной кислоты в растворе |
57 |
|
84 |
|
|
3.5.3. Определение солей аммония и азота в органических |
|
|
соединениях по Кьельдалю |
59 |
|
Глава 4. Окислительно-восстановительное титрование |
|
|
(редоксиметрия) |
61 |
|
4.1. Общая характеристика редокс-методов |
61 |
|
4.2. Перманганатометрия |
63 |
|
4.3. Дихроматометрия |
67 |
|
4.4. Иодометрия |
68 |
|
Глава 5. Методы осаждения и комплексообразования |
74 |
|
5.1. Осадительное титрование |
74 |
|
5.1.1. Аргентометрическое титрование |
74 |
|
5.1.2. Муркурометрическое титрование |
77 |
|
5.2. Комплексиметрические титрование |
77 |
|
5.2.1. Разновидности комплексиметрии |
77 |
|
5.2.2. Комплексонометрия |
78 |
|
Библиографический список |
82 |
Рассчитать навеску NaCl для приготовления 400мл 25% раствора NaCl.
Перед вами страница с вопросом Рассчитать навеску NaCl для приготовления 400мл 25% раствора NaCl?, который относится к
категории Химия. Уровень сложности соответствует учебной программе для
учащихся 5 – 9 классов. Здесь вы найдете не только правильный ответ, но и
сможете ознакомиться с вариантами пользователей, а также обсудить тему и
выбрать подходящую версию. Если среди найденных ответов не окажется
варианта, полностью раскрывающего тему, воспользуйтесь «умным поиском»,
который откроет все похожие ответы, или создайте собственный вопрос, нажав
кнопку в верхней части страницы.
