Бензойная кислота,
C6H5COOH — средняя* одноосновная кислота,
диссоциация которой представлена следующими ступенчатыми реакциями c соответствующими ступенчатыми константами диссоциации**.
| `”H””B” ⇄ “H”^+ + “B”^(-1)`, | `k_1 = ([“H”^+][“B”^(-1)])/([“H””B”])`, | `”p”k_1=4.21` |
* по первой ступени диссоциации (или первой табулированной константе диссоциации)
** источник –
“Справочник по аналитической химии. Издание 4-е.”, Лурье Ю.Ю.. Москва. “Химия”, 1971. c.249
На графике представлена кривая титрования бензойной кислоты кислоты основанием, рассчитанная по формулам.
В правой колонке вы можете выбрать индикатор с требуемым интервалом pH перехода окраски.
На вкладке «Параметры расчета» вы можете поменять входные данные для расчета (концентрации, начальный объем), а также параметры отображения графика.
Вы можете также посмотреть диаграмму распределения кислотных форм бензойной кислоты кислоты в зависимости от pH раствора.
Иван Иванов
Ученик
(86),
закрыт
9 лет назад
Рассчитайте рН 1 М раствора бензойной С 6 Н 5 СООН кисло – ты, константа диссоциации которой равна 1,6? 10 n6 .
Лучший ответ
ᅟ
Высший разум
(217790)
9 лет назад
Для слабых кислот:
рН = 1/2 (pKa + pCa).
pKa = – lg Ka,
Ka – константа кислотности – константа диссоциации слабой кислоты.
pCa = – lg Cа,
Са – молярная концентрация кислоты.
Могу проверить Ваш расчёт.
.
Остальные ответы
Похожие вопросы
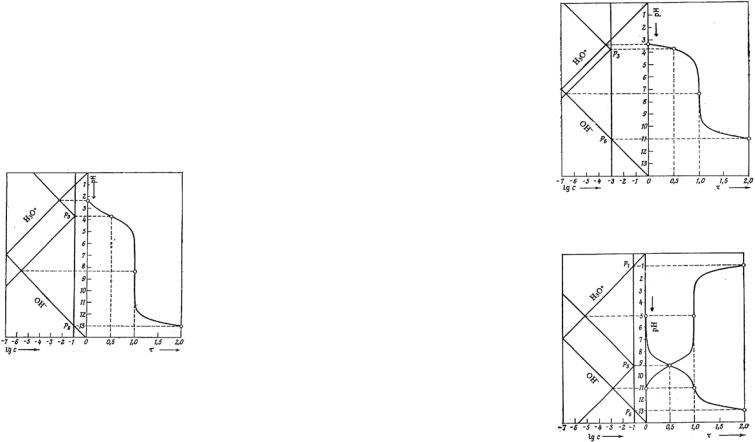
Из рассмотрения точек Р1-Р5 на рис.3.3 можно сделать еще некоторые выводы:
а) При смешении 0.1 моль С6Н5СООК и 0.1 моль НСl в воде до общего объема 1 дм3 раствора получают 0.1М раствор бензойной кислоты, содержащий КСl, рН которого определяют в точке Р2, где СА− = СН+. Это объясняется тем, что бензоат-ионы, являющиеся слабым основанием, полностью протонированы ионами гидроксония, обладающими сильнокислотными свойствами.
б) Раствору смеси бензойной кислоты и бензоата калия в эквимолярных соотношениях (т.е. слабой кислоты и сопряженного с ней основания) соответствует точка, где СНА = СА-, т.е. Р3, поэтому рН этого раствора равно рКа бензойной кислоты (независимо от концентрации кислотно-основной пары). Как было показано выше, при титровании это соответствует нейтрализации половины количества кислоты.
в) При растворении 0.1 моль С6Н5СООН и 0.1 моль КОН (объем раствора 1 дм3) получают 0.1М раствор бензоата калия, рН которого определяют в точке Р5, где СНА= СОН-. Таким образом, слабая бензойная кислота полностью депротонирована сильным основанием.
г) Для 1 дм3 раствора, содержащего cмесь 0.1 моль НСl и 0.1 моль бензойной кислоты, рН определяют в точке Р1, так как только в этой точке СН+ = СНА. В этом растворе значение рН такое же, как и в растворе индивидуальной сильной кислоты. Диссоциация слабой ки-
слоты полностью подавлена
HA ↔ H+ + A−
так как при добавлении сильной кислоты в виде ионов Н+ их избыток по принципу Ле-Шателье смещает равновесие данного процесса справа налево, т.е. в сторону образования недиссоциированных молекул слабой кислоты.
Этот вывод справедлив также для смеси децимолярных растворов гидроксида калия и бензоата калия, рН которой определяется в
точке Р6, где СОН– = СА-.
Используя соотношения, полученные из данных рис. 3.3, из уравнений (3.11) и (3.13) можно вывести полезные для практики формулы приближенного расчета рН растворов кислот и оснований.
Водные растворы кислот. Из уравнения (3.11) следует, что в растворе кислоты СН+ ≈ СА-. На рис.3.3 этому соответствует точка Р2, где СА– << СНА. Тогда уравнение (3.7) можно переписать как
СНА = С0 (3.14)
Подставив уравнения (3.11) и (3.14) в выражение закона действующих масс [уравнение (3.4)], получим для вычисления рН разбав-
101
ленной кислоты выражение (3.15), логарифмирование которого дает уравнение (3.16):
|
СН+2/С0 = Ка |
(3.15) |
|
рН = (1/2) (рКа – lg С0) |
(3.16) |
Водные растворы оснований. Выражение для расчета рН растворов оснований получим, используя соотношения, найденные из правой
части рис.3.3 (рН > 7): СА– >> СНА и СНА >> СН+. Вводя их в уравнения (3.7) и (3.12), получаем соответственно приближенные равенства
|
С0 ≈ СА– |
(3.17) |
|
СОН– ≈ СНА |
(3.18) |
Объединение уравнений (3.17) и (3.18) с выражением закона действующих масс [уравнение (3.4)] дает
|
СН+ С0/СОН– = СН+2 С0/Кw = Кa |
(3.19) |
|
рН = (1/2) (рКa + рКw + lgС0) |
(3.20) |
Общую концентрацию С можно определить как количество соли в молях, деленное на конечный объем раствора в дм3. В уравнении (3.20)
|
можно заменить рКa на рКb из соотношения рКa + рКb = рКw: |
|
|
рН = рКw – (1/2) (рКb – lgС0) |
(3.21) |
Уравнение (3.16) для расчета рН слабых кислот аналогично можно преобразовать в уравнение для расчета рОН слабых оснований, заме-
|
нив рН на рОН и рКa на рКb: |
|
|
рОН = (1/2) (рКb – lg С0) |
(3.22) |
Уравнение (3.22) идентично уравнению (3.21). Это наглядно видно, если выразить в уравнении (3.22) величину рОН через рН из условия pH + pOH = pKw
Уравнение (3.21) позволяет рассчитывать рН растворов, содержащих соль слабой кислоты и сильного основания. Такой раствор образуется также в точке эквивалентности при титровании слабой кислоты раствором сильного основания. Значение рН, рассчитанное по уравнению (3.21), называют показателем титрования рТ. Уравнения (3.16) и (3.21) справедливы только при соблюдении условий рис.3.3, т.е. для умеренно разбавленных растворов и для растворов кислот и оснований средней силы.
В табл.3.1 приведены формулы для расчета рН одноосновных кислот и границы применимости этих формул. Соответствующие формулы для расчета рН оснований получаются при замене СH+ на СOH– и
Кa на Кb.
Область применения уравнения (3.16) можно определить при сравнении логарифмической формы этого уравнения и рис.3.2, на котором уравнением (3.16) описываются нисходящие прямые с tg α = -2. Однако эти прямые только в области средних значений рК могли бы
102

перекрываться кривыми, рассчитанными из уравнения третьей степени. Уравнение (3.16), таким образом, может быть применимо только к сильным и очень слабым кислотам умеренных концентраций. Напротив, уравнение (3.23) (табл.3.1) применимо почти всегда и может давать отклонения только в области экстремально малых значений концентраций. Уравнение (3.23) перекрывает область действительных значений уравнения (3.16) и при Кa2/4 << КaС0 переходит в него. Уравнение (3.9) охватывает области применимости уравнений (3.16) и (3.23). При КwКa <СH+3 и КbС0 >>Кw оно переходит в уравнение (3.23,
табл.3.1).
С помощью диаграммы lgC-pH можно также определить концентрационные соотношения в растворе двух слабых кислот, например, иона аммония и уксусной кислоты (рис.3.4).
На диаграмме различают три области значений рН: рН< рКа1; рКа1 < рН<рКа2; рН>рКа2. В первой области обе кислоты NH4+ и CН3СООН находятся в недиссоциированном состоянии.
Рис.3.4. Логарифмическая рН-диаграмма для раствора 0.1М по уксусной кислоте и 0.1 М по NH4+
Таблица 3.1
Формулы для расчета рН растворов слабых кислот
|
Границы применимости |
Формула |
|||
|
Увеличение |
– >> |
– |
C |
+ = (K C )1/2 |
|
CA |
COH |
H |
a 0 |
|
|
разбавления |
CA– << CHA |
pH = (pKa– lgC0)/2 |
||
|
CA– >> COH– |
СH+=[(Ka2/4)+KaC0]1/2-Ka/2 |
|||
|
CA– ≈ CHA |
(3.23) |
|||
|
CA– ≈ COH– |
(CH+)3+Ka (CH+)2 – |
|||
|
↓ |
CA– ≈ CHA |
-(Kw+KaC0)CH+ – KwKa = 0 |
Во второй области в основном существует более слабая кислота NH4+, а также более слабое основание СН3СОО−, а в третьей области почти исключительно находятся основания NH3 и CH3СОО−.
Отсюда ясно, что при кислотно-основном титровании смеси двух слабых кислот происходит их последовательная депротонизация: вначале более сильной кислоты, а затем более слабой. Интересна точка Р. Для нее справедливо условие С(NH4+) ≈ C(СН3СОО−), которое выполняется и для раствора ацетата аммония, так что абсцисса точки Р соответствует значению рН этого раствора. Из рис.3.4 можно вывести выражение для точки Р, пригодное для расчета рН растворов солей, образованных анионами слабой кислоты и катионами слабого основания:
рН = (1/2) (рКa1 + рКa2 ) (3.24)
Оно показывает, что рН такого раствора определяется только значениями констант диссоциации обеих кислот и не зависит от концентрации.
Уравнение (3.24) можно упростить введением в него константы автопротолиза воды:
|
рН = 7 + (1/2) (рКa1 – рКb2 ) |
(3.25) |
Данное уравнение показывает, что если кислота 1 сильнее основания 2, то раствор соли, образованной катионом этого основания и анионом этой кислоты, имеет кислую реакцию; если же основание 2 сильнее кислоты 1, то раствор соли дает щелочную реакцию.
Уравнения (3.24) и (3.25) действительны, если выполняются неравенства СН+<<СА– и СОН-<< СНА , и недействительны для кислот или оснований, сила которых превышает определенное значение.
III.2.1. Математическое описание логарифмических рН-диаграмм
Кривые, приведенные на диаграмме lgC – pH, построены на основании уравнений, в которых концентрации СНА и СА– являются
функцией рН. Эти зависимости выведены комбинированием уравнений (3.4), (3.5), (3.6) и (3.7). При решении их относительно СНА и затем относительно СА– получают следующие уравнения:
|
СНА = С0СН+/(CH+ + Ka) = C0/(1+Ka/CH+) = C0/[1+10(pH-pKa)] |
(3.26) |
|
СA– = C0Ka/(CH++Ka) = C0/(1+CH+/Ka) = C0/[1+10(pKa-pH)] |
(3.27) |
|
Логарифмирование уравнения (3.26) приводит к (3.28) |
|
|
lgСHA = lgС0 – lg[1+10(pH -pKa)] |
(3.28) |
|
а уравнения (3.27) – к выражению (3.29) |
|
|
lgСA– = lgС0 – lg[1+10(pKa-pH)] |
(3.29) |
При графическом изображении этих зависимостей на диаграмме lgC0 –
pH получаются сильно растянутые изогнутые линии, представленные на рис.3.3. Их можно найти аппроксимацией двух областей каждой кривой. Для кривой НА (линии кислоты) используют:
|
а) область рН< рКа. При этом условии в уравнении (3.28) вели- |
|
|
чина 10(рН-рКа) будет намного меньше 1, откуда для левой ветви кривой: |
|
|
lgCНА = lgС0 |
(3.30) |
Это означает, что линия кислоты в области рН< рКa параллельна оси
|
рН и отстоит от нее на расстоянии lgC0. |
|
|
б) область рН> рК. В этом случае 10(pH−pKa) >>1, и поэтому из |
|
|
уравнения (3.28) следует |
|
|
lgCHA = pKa – pH + lgС0 |
(3.31) |
Эта функция представлена на диаграмме в виде нисходящей под углом 450 кривой (при одинаковом масштабе по осям ординат и абсцисс), пересекающей верхнюю кривую [уравнение (3.30)] при рН = рКa.
в) точку рН = рКa. Из уравнений (3.28) и (3.29) получаются зна-
|
чащие ординаты |
|
|
lgCHA = lgСA− = lgС0 − lg2 = lgС0 − 0.301 |
(3.32) |
|
Аналогично для линии основания используют: |
|
|
а) при рН< рКа из уравнения (3.29) величина следует |
|
|
lgCА− = pH − pKа + lgС0 |
(3.33) |
Уравнение (3.33) описывает левую прямолинейную ветвь линии основания, восходящую под углом 450.
б) область рН>рКa. При этом из уравнения (3.29) получают
Уравнение (3.34) описывает ветвь линии основания, параллельной оси рН.
|
в) точку рН = рКa. При этом, как было показано выше, |
|
|
lgCA− = lgС0 − 0.301 |
(3.35) |
III.3. МЕТОД НЕЙТРАЛИЗАЦИИ (КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ)
III.3.1. Кривые титрования
Зависимость значений рН, изменяющихся в процессе титрования, от добавляемого количества титранта графически изображается кривыми титрования, причем для простоты часто пренебрегают увеличением объема (разбавлением раствора). На оси ординат откладывают значения рН, на оси абсцисс – степень оттитровывания электролита r = С/Сэ, где С – количество добавляемого титранта в молях на кубический дециметр титруемого раствора, Сэ – количество титранта, необходимое
105
в соответствии с уравнением реакции, также в моль/дм3 раствора пробы. Обычно интерес представляет область 0<r<2, причем r = 0 соответствует чистому титруемому веществу до начала титрования, r = 0.5 – добавлению половины, r = 1 – добавлению всего и r = 2 – добавлению двойного количества титранта, рассчитанного из стехиометрических соотношений. При r = 1 получают точку эквивалентности.
Расчет интегральных кривых титрования
Титрование 100 мл 0.05 н. раствора сильной кислоты 0.1н. раствором сильного основания NaOH
Приводим расчет величин рН в различные моменты титрования, для экономии места не представляем рутинных расчетов рН во всех точках титрования, а покажем расчеты в характерные моменты титрования: в начале титрования, в момент полунейтрализации, в точке эквивалентности и после точки эквивалентности.
а) В начале титрования, когда еще титрант не был добавлен (V = 0 мл NaOH), концентрация ионов водорода в исходном растворе равна концентрации сильной кислоты, полностью продиссоциированной на
|
ионы водорода и хлорида: |
|
|
CHAn = [H+] = [Cl−] |
(3.36) |
|
т.е. [H+] = 5·10−2 моль/л или −lg(5·10−2) = 1.30. При добавлении сильно- |
го основания в качестве титрующего агента концентрация кислоты,
|
следовательно, концентрация ионов водорода уменьшается. |
|
|
б) Прилито 25 мл основания. При этом |
|
|
[H+] = (СэкHCl·VHCl − СэкNaOH·VNaOH)/(VHCl+VNaOH) |
(3.37) |
|
т.е. [H+] = (0.05·100−0.1·25)/(100+25) = 2·10−2 или рН = 1.70. |
|
|
в) Прилито 50 мл раствора основания. При этом наблюдается |
полная нейтрализация кислоты и величина рН в точке эквивалентности (рТ) равна рН = 7.00.
г) Прилито 51 мл титрующего агента. За точкой эквивалентности будет избыток титранта, а значит, ионов ОН−. Рассчитаем количество
|
избытка щелочи: |
|
|
[OH−] = (СэкNaOH·VNaOH − NHCl·VHCl)/(VHCl+VNaOH) |
(3.38) |
|
Тогда [OH−] = (51·0.1−100·0.05)/(51+100) = 6.62·10−4 моль/л |
[H+] = 10−14/6.62·104 = 1.51·10−11 или рН = 10.82.
Обычно по полученным данным (объем титранта и рН раствора) строят интегральную кривую титрования в координатах рН – V, определяют объем титранта в момент эквивалентности и проводят дальнейшие расчеты. А на практике количество кислоты не бывает извест-
106
ным, поэтому для фиксирования точки эквивалентности и расчетов по определению концентрации кислоты применяют соответствующий кислотно-основный индикатор, изменяющий свою окраску в точке эквивалентности.
Титрование 100 мл 0.05н. раствора уксусной кислоты 0.1н. раствором гидроксида натрия
а) VNaOH = 0 мл. Тогда рН до начала титрования определяется концентрацией (CHan) и константой диссоциации (KHan = 1.8·10−5) слабой уксусной кислоты:
|
[H+] = (KHan·CHan)1/2 |
(3.39) |
тогда [H+] = (1.8·10−5·5·10−2)1/2 = 9.49·10−4 моль/л или величина рН = 3.02.
б) VNaOH = 25 мл. Как только начали титрование, приливая небольшими порциями щелочь, в растворе наряду с остающейся в избытке до момента эквивалентности слабой кислотой образуется ацетат натрия переменного количества (количество свободной кислоты непрерывно уменьшается, тогда как количество образующегося ацетата натрия – продукта взаимодействия титранта с титруемым веществом − увеличивается). Другими словами, в титруемом растворе образуется ацетатная буферная смесь, рН которой при изменении объема титранта до точки эквивалентности рассчитывается по формуле:
[H+] = KHАn·CHАn/CKtAn (3.40)
Количества оставшейся кислоты и получающейся соли (ацетата натрия) до точки эквивалентности можно определить следующими уравнениями:
CHan = (СэкHАn·VHАn − СэкNaOH·VNaOH)/(VHАn+VNaOH), CKtAn = (СэкNaOH·VNaOH)/(VHАn+VNaOH),
т.е. количество моль эквивалентов оставшейся слабой кислоты равно разности исходного количества моль эквивалентов кислоты и количества моль эквивалентов кислоты, перешедшей в результате реакции нейтрализации в ацетат натрия и равной количеству моль эквивалентов добавленного основания. Итак, при добавлении 25 мл титранта
CHАn = (0.05·100 − 0.1·25)/(100+25) = 2·10−2 моль/л,
CKtAn = 0.1·25/(100+25) = 2·10−2 моль/л.
Тогда [H+] = 1.8·10−5 ·(2·10−2/2·10−2) = 1.8·10−5 моль/л, а величина рН = 4.74.
в) VNaOH = 50 мл. Кислота полностью нейтрализована, в титруемом растворе уже нет свободной уксусной кислоты, но еще нет и избытка основания, все количество кислоты перешло в столько же моль
эквивалентов соли слабой кислоты и сильного основания, тогда в этой точке имеется раствор соли CH3COONa, подвергающейся гидролизу по аниону. Величину рН в точке эквивалентности (рТ) можно рассчитать по соответствующей формуле (для соли, подвергающейся гидролизу по аниону):
|
[H+] = (Kw·KHАn/CKtAn)1/2 |
(3.41) |
Здесь Kw − ионное произведение воды (константа автопротолиза растворителя); KHАn − константа диссоциации слабой уксусной кислоты; CKtAn − концентрация соли CH3COONa, равная числу моль эквивалентов исходной кислоты или основания в точке эквивалентности
[(СэкHan·VHan)/(VHan+VNaOH) = (СэкNaOH·VNaOH)/(VHan+VNaOH)].
[H+] = [(10−14·1.8·10−5)/(10−1·50/150)]1/2 = 2.32·10−9 моль/л или рН = 8.63.
г) Прилито 51 мл 0.1 н. раствора NaOH. В титруемом растворе имеется некоторый избыток щелочи, приводящий к величине рН > 7. Рассчитаем соответствующее значение рН в этой точке по избытку ионов ОН−. Избыток гидроксид-ионов находим по количеству щелочи за точкой эквивалентности:
[OH−] = (СэкNaOH·VNaOH − NHan·VHan)/(VHan+VNaOH) =
= (0.1·51 −0.05·100)/(100+51) = 6.62·10−4 [H+] = 1·10−14/6.62·10−4 = 1.51·10−11 моль/л или рН = 10.82.
Таким образом, по величинам рН, рассчитанным в каждой точке титрования, в зависимости от объема титранта можно было бы построить кривые титрования кислот разной силы, т.е. HCl и более слабых кислот с различными значениями Кдисс.
При построении зависимости значений рН от объема титранта или лучше от степени оттитровывания в ходе титрования кислот различной силы и равной концентрации раствором сильного основания получают кривые, изображенные на рис. 3.5.
Из этих кривых можно сделать некоторые важные общие выво-
ды.
1. Чем слабее кислота, тем больше изменение рН при добавлении первых капель раствора основания. Это видно, если на рис.3.5 нанести значения рН раствора чистой кислоты, полученные из рис.3.2 или из соответствующей диаграммы lgC-pH. Кривая с рК = 8 “выпрыгивает” из ординаты при рН = 5.5, кривая с рК = 4 – при рН = 2.5, а кривая сильной кислоты – при рН = 1.
2. Кривая титрования слабой кислоты несимметрична относительно линии нейтральности (рН = 7) и рТ в точке эквивалентности

находится выше линии нейтральности, причем чем слабее кислота, тем асимметричнее кривая титрования или тем величина рТ > 7.
Рис.3.5. Кривые титрования 0.1 н. растворов кислот с различными значениями констант кислотности 0.1 н. раствором сильного основания
3. На кривых титрования слабых кислот в точке полунейтрализации (т.е. при степени оттитровывания r = 0.5) рН = рК.
То же самое можно вывести из выражения для закона действующих масс после решения уравнения относительно СН+ и логарифмирования:
|
pH = pKа – lg(СA-/СHA) |
(3.42) |
При добавлении половины стехиометрического количества раствора основания, т.е. при r = 0.5, с достаточным приближением можно считать СA– = СHA и тогда получим lg(СA-/СHA) = 0. При этом уравнение
(3.42) превращается в (рН1/2 = рКa), где рН1/2 – это рН при r = 0.5. Возрастающий полого участок кривой, в середине которого на-
ходится рН1/2, характеризуется тем, что даже при добавлении относительно больших количеств титранта – основания – значение рН изменяется незначительно. Такое поведение называется буферным действием, а соответствующий раствор – буферным раствором. В нашем случае буферная смесь состоит из слабой уксусной кислоты и ацетата натрия.
4. Так называемый показатель титрования рТ, т.е. рН в точке эквивалентности, как отмечалось чуть раньше, тем дальше сдвинут в основную область, чем слабее титруемая кислота. При этом скачок рН в области точки эквивалентности увеличивается с увеличением силы
109
титруемой кислоты. Это явление имеет большое практическое значение: чем меньше скачок титрования, тем менее круто восходит кривая титрования в области точки эквивалентности и тем менее точно можно определить точку перегиба и объем титранта в точке эквивиалентности. Поскольку точку эквивалентности при титровании определяют непосредственно по абсциссе точки перегиба на кривой титрования, результаты титрования в целом тем менее точны, чем слабее титруемая кислота. Очень слабые кислоты с константами диссоциации Кдисс < 10−9 не титруются в водных средах. Показатель титрования слабой кислоты можно определить из диаграммы lgC – pH (точка Р5 на рис.3.3) или из кривой сопряженного с кислотой основания, представленной на рис.3.2.
Рис.3.6. Кривые титрования сильных и слабой кислот различной концентрации сильным основанием
Пример. Сопряженное с 0.1н кислотой, рКа которой равно 8, такой же концентрации (0.1н.) основание имеет рКb = 6, так как рКa + рКb = 14, и соответствующее ему значение рН равно 10.5 (рис.3.2).
На рис.3.6 показано влияние исходной концентрации титруемой кислоты на ход кривой титрования слабой или соответственно сильной кислоты. Из рис.3.6 можно сделать следующие выводы:
а) Скачок рН в области точки эквивалентности при титровании сильной кислоты тем меньше, чем меньше исходная концентрация кислоты.
б) Ход кривых титрования слабых кислот в области 0 < r < 1 в меньшей степени зависит от исходной концентрации кислот, чем ход кривых титрования сильных кислот. Кривые титрования слабых кислот
110
проходят (за исключением кривых для концентраций С0 < Ка) через точку, в которой r = 0.5, рН =рКа.
III.3.2. Связь между диаграммами lgC-pH и кривыми титрования
Логарифмическая диаграмма особенно упрощает построение кривой титрования, как это видно из рис.3.7 на примере титрования слабой кислоты раствором сильного основания. На рис.3.8 и 3.9 представлены другие типы диаграмм – для особенно низких исходных концентраций и для очень слабых кислот, соответственно.
Рис.3.7. Логарифмическая рН-диаграмма и кривая титрования для 0.1 н. раствора муравьиной кислоты сильным основанием
Такие комбинированные диаграммы дают возможность получать дополнительную информацию из диаграмм lgC−pH. На рис.3.9 приведена комбинированная диаграмма кислотно-основной пары NH4+/NH3 при концентрации 0.1 моль/дм3.
Из формы обеих кривых ясно, что титрование аммиака соляной кислотой характеризуется большей точностью, чем титрование хлорида аммония раствором едкого натра. Так, длина отрезка Р3Р6 (рис.3.8) является достаточно точной мерой скачка рН при титровании кислоты раствором сильного основания, а длина отрезка Р3Р1 дает величине скачка при титровании сопряженного основания раствором сильной кислоты.
Рис.3.8. Логарифмическая рН-диаграмма и кривая титрования для 0.001 н. раствора муравьиной кислоты сильным основанием
Рис.3.9. Логарифмическая рН-диаграмма и кривая титрования для 0.1 н. раствора NH4Сl или раствора NH3 cоответственно
Из диаграммы lgC-pH можно также построить кривую дифференцированного титрования кислот в их смеси. На рис.3.10 приведена диаграмма для раствора, содержащего смесь соляной (хлороводородной) и уксусной кислот с концентрацией каждой 0.1 моль/дм3, а на рис.3.11 – диаграмма для раствора смеси двух слабых кислот с рКа1 = 3.15 и рКа2 = 6.9.

Рис.3.10. Логарифмическая рН-диаграмма и кривая титрования для смеси хлористоводородной и уксусной кислот (С0 = 0.1 моль/ дм3). Титрант – сильное основание (С = 0.1 моль/ дм3)
Рис.3.11. Логарифмическая рН-диаграмма и кривая титрования для раствора двух кислот с рКа1 = 3.15 и рКа2 = 6.9 (С0 = 0.1 моль/дм3)
Из рис.3.11 ясно, что кислоты в их смеси можно оттитровать дифференцированно с достаточной точностью, если значения рКа отличаются по меньшей мере на 3 единицы.
На рис.3.12 приведена комбинированная диаграмма для фосфорной кислоты H3РО4. При титровании H3РО4 проявляет свойства двухосновной кислоты, поскольку третья константа диссоциации (рКа3 = 12.66) настолько смещена в основную область, что по третьей ступе-
113
ни кислоту нельзя оттитровать обычным путем (т.е. прямым титрованием).
Рис.3.12. Логарифмическая рН-диаграмма и кривая титрования для фосфорной кислоты (рКа1 = 2.2, рКа2 = 7.2 и рКа3 = 12.66; С0 = 0.1 моль/дм3). Титрант – сильное основание
III.4. Кислотно-основные индикаторы
III.4.1. Теории индикаторов
Как отмечалось несколько ранее, для возможно точного определения количества вещества (кислоты, основания, различных солей) в анализируемой пробе (в навеске или в данном объеме) при кислотноосновном титровании необходимо, чтобы в момент эквивалентности (в точке эквивалентности) окраска раствора изменилась. Этого можно достичь при применении кислотно-основных индикаторов, изменяющих свою окраску при рН в конечной точке титрования.
Индикаторы, применяемые в методе нейтрализации, представляют собой органические вещества достаточно сложного строения, принадлежащие к различным классам органических соединений. Все кислотно-основные индикаторы обладают одним общим свойством – они изменяют свою окраску в зависимости от изменения рН раствора.
III.4.1.1. Теория индикаторов Оствальда
Эта теория называется ионной теорией индикаторов.
114
В ее основу положена теория электролитической диссоциации. 1. Все индикаторы – слабые электролиты, обладающие кислыми и основными свойствами. Индикаторы, способные отдавать свои
протоны, называются кислотными индикаторами и обозначаются
HInd.
HInd ↔ H+ + Ind−
донор протона
Индикаторы же, способные присоединять протоны, называются основными и обозначаются IndOH, IndNH2 и т.д. :
IndOH ↔ Ind+ + OH−
акцептор протона
В обоих случаях Ind− и Ind+ – ионная форма соответствующего индикатора.
Поэтому индикаторы, применяемые в методе кислотноосновного титрования, носят название кислотно-основных индика-
торов.
2. Индикаторы метода нейтрализации (с переносом протона)
обладают различной окраской в молекулярной и ионной формах.
3.Изменение окраски индикатора является обратимым процес-
сом.
Основные положения теории
Как уже отмечалось, индикаторы являются электролитами и диссоциируют в воде согласно уравнениям:
|
HInd |
↔ H+ + Ind− |
(а) |
|
1 окраска |
2 окраска |
|
|
IndOH |
↔ Ind+ + OH− |
(б) |
|
1 окраска |
2 окраска |
В водном растворе индикатор – кислота (а) – диссоциирует с выделением катиона Н+ и аниона Ind−. При растворении в воде устанавливается динамическое равновесие диссоциированных и недиссоциированных молекул индикатора. И если к этому раствору прилить раствор кислоты, то происходит увеличение концентрации ионов водорода и равновесие (а) сдвигается влево (←), тем самым увеличивая концентрацию недиссоциированных молекул и раствор примет 1 окраску. Если же прилить к раствору, содержащему индикатор, раствор щелочи, то появление в растворе гидроксид-ионов вызывает связывание ионов Н+ в слабо диссоциированные молекулы воды, что приводит к смещению равновесия вправо (→):
115
HInd ↔ H+ + Ind−
+
ОН−
↓
Н2О
В растворе увеличивается концентрация ионов Ind− и раствор примет 2 окраску, при этом индикатор диссоциирует полнее. Константу электролитической диссоциации индикатора представим в виде:
Кинд = [H+]·[Ind−]/[HInd]
Так как Кинд является величиной постоянной, то прибавление в раствор кислоты ведет к увеличению [H+] и [НInd] и уменьшению [Ind−], т.е. к усилению окраски, свойственной молекулярной форме индикатора. Прибавление в раствор щелочи вызывает обратное действие – усиление окраски, свойственной ионной форме индикатора.
Таким образом, переход одной окраски, присущей молекулярной форме кислотно-основного индикатора, в другую свойственную его ионной форме, зависит от величины рН раствора.
Теория Оствальда не была лишена недостатков и не давала объяснения следующим явлениям:
1)изменение окраски индикаторов происходит не мгновенно, что свойственно ионной реакции, а протекает несколько медленнее;
2)изменение окраски индикаторов связано с изменением структуры их молекул – внутримолекулярной перегруппировкой, вызывае-
мой действием ионов Н+ и ОН−.
III.4.1.2. Хромофорная теория индикаторов
Основные положения этой концепции:
1) Окраска органических соединений (а кислотно-основные индикаторы представляют собой именно органические соединения) обусловлена наличием в их молекулах определенных групп, называемых
хромофорами:
азогруппа: −N=N−
нитрозогруппа: −N=O
нитрогруппа: −NO2
хиноидная структура (группа): =С6Н4=
Помимо этих групп в индикаторах обычно присутствуют группы, называемые ауксохромами (−OH, −NH2, −NHR, −NR2). Эти группы сообщают соединениям кислотный или основный характер (помимо этого они способствуют углублению окраски индикатора).
116
2) Каждый индикатор существует в двух формах (А и В), которые находятся в равновесии друг с другом и могут переходить одна в другую:
HInd ↔ HInd
A B
Формы А и В имеют различное строение и потому имеют различную окраску. Эти формы называются таутомерными (говорят, что обе формулы представляют одно и то же химическое соединение в различных таутомерных формах).
3) Различные таутомерные формы индикаторов имеют различную окраску индикаторов, меняющуюся при действии кислот и оснований. Это можно объяснить тем, что форма А индикатора не может диссоциировать, а 2 форма (В) индикатора может диссоциировать с образованием ионов Н+ и Ind−:
(HInd)A ↔ (HInd)B ↔ H+ + Ind−
Применив закон действующих масс (ЗДМ) к диссоциации формы В, можно записать:
K(Hind)B = [H+[·[Ind−]/[HInd]B
Но между двумя формами существует равновесие, которое подчиняется ЗДМ:
[HInd]B/[Hind]A = Kтаут
В большинстве случаев индикатор находится в форме А, а количество формы В – мало и его окраска незаметна. При добавлении щелочи (ОН−-ионов) концентрация [H+] понижается и усиливается диссоциация [HInd]B для постоянства KHind, в результате этого уменьшается концентрация [HInd]B.
III.4.1.3. Ионно-хромофорная теория индикаторов
Согласно дополняющим друг друга ионной и хромофорной теориям в растворах кислотно-основных индикаторов одновременно имеют место равновесия, обусловленные диссоциацией молекул и равновесия, связанные с внутримолекулярными перегруппировками одних форм индикатора в другие, отличающиеся различным строением. Поэтому обе рассмотренные теории объединены в одну общую ионнохромофорную теорию индикаторов.
Рассмотрим наиболее часто применяемые индикаторы кислотноосновного титрования, переход их окраски.
1. Фенолфталеин. Данный индикатор представляет собой бесцветное кристаллическое, нерастворимое в воде, но хорошо растворимое в спирте вещество. Поэтому в химических лабораториях применя-
ют 0.1% раствор индикатора в 50% этиловом спирте. В нейтральной и кислой среде фенолфталеин находится в так называемой лактонной форме, не имеющей окраски и имеет следующую структурную формулу:
OН
C
OН
O C O
бесцветная форма фенолфталеина
При действии на фенолфталеин разбавленных растворов щелочи, например NaOH, образуется двунатриевая соль, имеющая хиноидное строение, имеющая характерное окрашивание, присущее данному хромофору и окрашенное в красный цвет:
ONa
C
O
O  C O Na
C O Na
окрашенная форма фенолфталеина
В результате реакции происходит внутримолекулярная перегруппировка, показанная на нижеприведенной схеме:
Бесцветная лактонная форма Окрашенная хиноидная форма
|
2- |
|||||
|
OH |
OH – |
O |
|||
|
C |
C |
||||
|
OH |
H+ |
+ 2H2O |
|||
|
O |
|||||
|
O C O |
O |
C O |
или в более общей форме:
фН + NaOH ↔ фNa + H2O
Хиноидная группа присутствует и в нейтральной, и в кислой среде, но в малых количествах, не превышающих 10%. Эти структурные изменения фенолфталеина (бесцветная лактонная форма – окрашенная хиноидная форма) наблюдаются при значениях рН = 8…10. Но при дальнейшем увеличении рН раствора (при добавлении избытка щелочи, рН = 13…14) фенолфталеин претерпевает новую структурную перегруппировку. При этом образуется тризамещенная натриевая соль, не имеющая хиноидного строения:
OH
ONa
C
ONa
O  C ONa
C ONa
2. Метиловый оранжевый (гелиантин, натриевая соль N- диметиламинобензолсульфокислоты) – один из наиболее часто используемых в химических лабораториях индикатор, представляет собой растворимый в воде оранжево-желтый порошок. В химической лаборатории обычно применяется 0.05% водный раствор. Имеет следующую структурную формулу:
|
(CH3)2 N |
N N |
SO3– |
H+ |
|
OH– |
|||
|
(CH3)2 |
N |
N N |
SO3– |
Отметим, что в верхней части (в нейтральной и щелочной среде) окраска индикатора в азоформе – желтая, в то время как при рН < 3.1 (нижняя часть структурной формулы) индикатор принимает хиноидную форму, окрашенную в красный цвет.
В первом случае при рН > 4.4 в молекуле индикатора метиловый оранжевый хромофорной группой является азогруппа –N=N−, которой свойственна желтая окраска, и ауксохромная сульфогруппа −SO3H, углубляющая окраску раствора.
При добавлении к раствору с индикатором кислоты (при уменьшении рН <3.1) происходит изменение структуры молекулы индикатора – в молекуле появляется хиноидная группа
3. Метиловый красный. Молекула метилового красного по своей структуре в кислой и щелочной средах практически ничем не отличается от молекулы метилового оранжевого. Они имеют лишь различные ауксохромные группы. Так, у метилового красного ауксохромной группой является −COONa. Добавим лишь, что введение ауксохромной группы −COONa в молекулу метилового красного приводит к смещению интервала перехода окраски индикатора в более основную область рН (до рН = 4.4 и более кислом растворе индикатор имеет красную окраску, а при рН, превышающих 6.2, раствор с индикатором метиловый красный принимает желтую окраску), так как заместитель СОО− является менее кислым, чем сульфогруппа SO32−.
Как отмечалось ранее, кислотно-основные индикаторы (рНиндикаторы) обычно представляют собой слабые органические кислоты или основания, окраска ионной и молекулярной форм которых раз-
лична. Если ионная и молекулярная формы абсорбируют свет в видимой области спектра, такие индикаторы называются двухцветными. Если только одна форма индикатора поглощает свет в видимой области спектра, то такой индикатор называют одноцветным (например, фе-
нолфталеин).
Двухцветные индикаторы. Реакцию нейтрализации индикатора, являющегося кислотой, можно представить следующим образом:
молекулярная ионная форма форма
Логарифмическое выражение закона действующих масс для этой реакции:
|
pH = pKHInd – lg(СHInd/СInd–) – lg(fHInd/fInd–), |
(3.44) |
где KHInd – константа диссоциации индикатора HInd; CHInd, fHInd, СInd–, fInd– – концентрация и коэффициент активности молекулярной и ионной
форм индикатора.
В разбавленном растворе принимают fHInd ≈ fInd–. Тогда каждому значению рН соответствует определенное соотношение СHInd/СInd–.
Если принять, что растворы с одинаковой концентрацией HInd и Ind– имеют одинаковую интенсивность окраски, которая пропорциональна концентрации, следует ожидать, что в пределах относительно большой области рН окраска ионной формы индикатора непрерывно переходит в окраску недиссоциированной молекулярной формы.
При этом было бы невозможно определить значение рН, так как нет резкого изменения окраски. Однако здесь сказывается невысокая чувствительность зрения человека: глаз до тех пор воспринимает, как было установлено ранее, окраску как чистую, пока содержание второго компонента в 10 раз не превысит содержание первого.
Чистую окраску второго компонента можно видеть, если ее интенсивность примерно в 10 раз больше, чем интенсивность окраски первого.
При переходе из кислой в щелочную область значение рН, при котором происходит первое видимое изменение окраски индикаторакислоты, можно определить на основании уравнения (3.41) по формуле
(рН)1 = рКHInd − lg10 = pKHInd − 1 (3.45)
При дальнейшем увеличении рН чистая окраска анионов индикатора возникает при
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
