Хроматография (ОФС.1.2.1.2.0001.15)
Государственная фармакопея 13 издание (ГФ XIII)
Хроматография — метод разделения смесей веществ, основанный на их многократном перераспределении между двумя контактирующими фазами, одна из которых неподвижна, а другая имеет постоянное направление движения.
ОБЩАЯ ФАРМАКОПЕЙНАЯ СТАТЬЯ
Взамен ст. ГФ XI, вып.1
Хроматографией называется метод разделения смесей веществ, основанный на их многократном перераспределении между двумя контактирующими фазами, одна из которых неподвижна, а другая имеет постоянное направление движения. По механизму, лежащему в основе разделения, различают адсорбционную, распределительную, ионообменную и другие виды хроматографии.
В настоящее время используются следующие хроматографические методы анализа, представленные на рис.1
Рисунок 1. Методы хроматографического анализа.

Результат хроматографического разделения представляется в виде хроматограммы.
ХРОМАТОГРАММА И ХРОМАТОГРАФИЧЕСКИЕ ПАРАМЕТРЫ
Хроматограмма представляет собой графическое или иное представление сигнала детектора, концентрации веществ в элюате или другой количественной величины, используемой для измерения концентрации веществ в элюате, от времени или объема подвижной фазы. В планарной (плоскостной) хроматографии хроматограммой называют также зафиксированную на бумаге (бумажная хроматография) или ТСХ-пластинке (тонкослойная хроматография) последовательность зон адсорбции веществ исходной (анализируемой) смеси.
Схематически хроматограммы представляют собой последовательность гауссовых пиков на базовой линии (рис. 2).
Базовая линия – сигнал от подвижной фазы.
Пик – часть хроматограммы, регистрирующая отклик детектора. Пик отображает постепенное нарастание концентрации вещества и последующее ее уменьшение. В случае линейной изотермы сорбции кривая, описывающая пик, приближается к кривой гауссова распределения.
Основание пика — продолжение базовой линии, соединяющее начало и конец пика.
Площадь пика (S) – площадь хроматограммы, заключенная между кривой, описывающей пик, и его основанием.
Высота пика (H) – расстояние от максимума пика до его основания, измеренное параллельно оси отклика детектора.

Рисунок 2. Хроматограмма и основные хроматографические параметры: 1 и 2 – пики соединений 1 и 2; t1 и t2 – соответствующие времена удерживания; t0 – время удерживания несорбирующегося вещества; W1 и W2 – ширина пиков у основания; W0,5 – ширина пика на половине его высоты (предполагается гауссова форма пиков)
Интерпретация хроматографических данных
Время удерживания (tR или t) – время, необходимое для элюирования вещества. Соответствует времени появления максимума пика на хроматограмме.
Объем удерживания (VR) – объем подвижной фазы, необходимый для элюирования вещества. Может быть вычислен по времени удерживания и скорости потока (F).
VR = F · tR
Объем удерживания, в отличие от времени удерживания, не зависит от скорости потока.
Время удерживания несорбирующегося вещества (t0 или tм) – время, необходимое для элюирования неудерживаемого на сорбенте вещества. В эксклюзионной хроматографии t0 соответствует времени удерживания веществ, размер молекул которых больше, чем наибольшие поры сорбента.
Объем удерживания несорбирующегося вещества (V0) – объем подвижной фазы, необходимый для элюирования неудерживаемого вещества. Может быть вычислен по времени удерживания неудерживаемого вещества и скорости потока (F):
V0 = F · t0
В эксклюзионной хроматографии V0 соответствует объему удерживания веществ, размер молекул которых больше, чем наибольшие поры сорбента.
Общее время удерживания подвижной фазы (tt) – в эксклюзионной хроматографии время удерживания веществ, молекулы которых меньше, чем наименьшие поры сорбента.
Общий объем удерживания подвижной фазы (Vt) – в эксклюзионной хроматографии объем удерживания веществ, молекулы которых меньше, чем наименьшие поры сорбента.
Константа (коэффициент) распределения (K0) – в эксклюзионной хроматографии характеристика элюирования вещества из определенной колонки, которую рассчитывают с помощью выражения:

Приведенное (исправленное) время удерживания вещества (t´R) ‒ время удерживания вещества за вычетом времени удерживания несорбируемого вещества. Может быть рассчитано по формуле:
t´R = tR — t0
Исправленное время удерживания не зависит от объема трубопроводов хроматографической системы, установленных между инжектором и колонкой.
Относительное время удерживания (r) – относительное приведенное (исправленное) время удерживания вещества 2 по веществу 1:

Нескорректированное относительное время удерживания (RRT) – относительное время удерживания вещества 2 по веществу 1:

Если не указано иное, значения относительного времени удерживания, приведенные в фармакопейных статьях, соответствуют нескорректированному относительному времени удерживания
Коэффициент емкости (k´) – коэффициент емкости колонки по веществу с временем удерживания tR, показывающий, во сколько раз исправленное время удерживания вещества больше, чем время удерживания несорбируемого вещества.

Эффективность хроматографической системы ‒ параметр, характеризующий степень размывания хроматографического пика. Эффективность выражается числом теоретических тарелок (N):

W – ширина пика у основания;
W0,5 – ширина пика на половине высоты.
При расчете числа теоретических тарелок значения времени удерживания и ширины пика должны быть приведены к одинаковой размерности.
Число теоретических тарелок зависит от природы определяемого вещества, его концентрации или объема, вводимого в систему, от колонки, температуры колонки и состава подвижной фазы.
Если не указано иное, то эффективность хроматографической системы, требования к которой приведено в фармакопейной статье, рассчитывается по формуле, использующей ширину пика на половине его высоты.
Фактор асимметрии (фактор симметрии) пика (As) рассчитывают по формуле:
 где
где
W0,05 – ширина пика на 5 % (1/20) его высоты;
f – расстояние между перпендикуляром, опущенным из вершины пика, и восходящей стороной пика на 5 % его высоты (рис. 3).
Если фактор асимметрии равен 1, то пик симметричен. Если фактор асимметрии больше 1, то это означает, что растянут задний фронт пика. Если фактор асимметрии меньше 1, то пик растянут спереди.

Рисунок 3. Схема расчета фактора асимметрии пика
Разрешение (Rs).
Разрешение между пиками двух веществ смеси элюирующимися друг за другом рассчитывают по формулам:

при этом tR2 ≥ tR1. При расчете разрешения величины времени удерживания и ширины пиков должны быть приведены к одинаковой размерности.
Если не указано иное, то разрешение, требования к которому приведены в фармакопейной статье, рассчитывается по формуле, использующей ширину пиков на половине высоты.
В случае, если пики несимметричны и если интенсивность пиков значительно различается, параметр Rs не всегда корректно описывает разделение хроматографических пиков. Таким образом, даже при значениях Rs ≥ 1,5 может наблюдаться неполное разделение пиков. В этих случаях при оценке разделяющей способности можно заменить параметр Rs на параметр «отношение максимум/минимум».
Отношение максимум/минимум (p/v), называемое также отношением «peak-to-valley», «пик – долина». Этот параметр позволяет оценить разделительную способность хроматографической системы. Значение p/v рассчитывается по формуле:
p/v = Hp / Hv
Hp – высота меньшего пика относительно экстраполированной базовой линии;
Hv – высота низшей точки (седловины) кривой, разделяющей пики, относительно экстраполированной базовой линии (рис. 4).

Рисунок 4. Хроматограмма не полностью разделяемых веществ
Данное соотношение применяется для оценки разделительной способности хроматографической системы, если вещества смеси разделяются не полностью. Рассчитанное отношение максимум/минимум в значительной степени зависит от выбранного варианта интегрирования хроматограммы. Результаты измерения соотношения p/v будут некорректны в случае разметки меньшего пика методом экстраполяции смещенной базовой линии (методом тангенциальной касательной).
Отношение сигнал/шум (S/N).
Краткосрочный шум сигнала детектора (шум базовой линии) влияет на прецизионность количественного определения. Отношение сигнал/шум рассчитывают по формуле:

Н – высота пика, соответствующего рассматриваемому веществу на хроматограмме указываемого стандартного раствора, измеренная от максимума пика до экстраполированной базовой линии. Экстраполяция базовой линии проводится для сигнала на участке базовой линии во временном интервале, продолжительность которого не менее 5-кратного значения ширины пика на его полувысоте;
h – размах фонового шума, измеряемый либо на хроматограмме контрольного (холостого) раствора (или раствора плацебо), либо на хроматограмме того же раствора стандартного образца.
Измерение размаха фонового шума проводится во временном интервале, продолжительность которого не менее 5-кратного значения ширины пика на его полувысоте, расположенном, если это возможно, равномерно по обе стороны от места возможного обнаружения пика. В случае использования для измерения шума хроматограммы контрольного (холостого) раствора (или раствора плацебо) измерение размаха фонового шума проводится во временном интервале, включающем в себя время удерживания рассматриваемого вещества, при этом продолжительность временного интервала, в котором проводится измерение шума, должна не менее чем в 5 раз превышать ширину на половине высоты для пика на хроматограмме указываемого раствора стандартного образца (рис. 5).

Рисунок 5. Вычисление отношения сигнал/шум.
Объем задержки (D) (объем задержки градиента) представляет собой объем системы между точкой, в которой происходит смешение элюентов, и началом колонки.
Объем задержки градиента влияет на времена удерживания веществ и общий профиль наблюдаемой хроматографической картины, получаемой при использовании градиентного элюирования. Объем задержки хроматографической системы определяется следующим способом.
Колонка. Заменяют хроматографическую колонку капилляром (например, 1 м × 0,12 мм).
Подвижная фаза.
Подвижная фаза A — вода.
Подвижная фаза B — 0,1 % (об/об) раствор ацетона.
Скорость потока. Необходимая для создания давления, достаточного для стабильной работы насоса (например, 2 мл/мин).

Детектор. Спектрофотометрический при длине волны 265 нм.
Определяют время (t0,5) в минутах, когда оптическая плотность увеличилась на 50 %. Вычисляют объем задержки:


Рисунок 6. Определение объема задержки градиента
Прецизионность системы в условиях повторяемости
Прецизионность системы в условиях повторяемости выражается в виде рассчитанного относительного стандартного отклонения в процентах [RSD (%)] по результатам последовательных измерений не менее чем 3 вколов или нанесений раствора сравнения и рассчитывается по формуле:

В планарной хроматографии аналогом времени удерживания является фактор удерживания (Rf):
Rf = a / b
где a – расстояние от точки нанесения пробы до центра пятна, характеризующего зону адсорбции;
b – расстояние от линии старта до линии фронта элюента.
На экспериментально определяемые значения Rf заметно влияют условия хроматографирования. Оценкой хроматографической подвижности, менее чувствительной к влиянию отклонений в условиях проведения эксперимента, является величина Rst, представляющая собой отношение величины Rf одного вещества к величине Rf другого, принятого за стандарт:


Рисунок 7. Схема определения значений Rf и Rst.
Данные планарной хроматографии могут быть представлены в виде денситограмм.
РАСЧЕТ СОДЕРЖАНИЯ ОПРЕДЕЛЯЕМЫХ ВЕЩЕСТВ
При расчетах содержания определяемых веществ пики растворителей и реактивов, подвижной фазы или среды (матрицы) образца не учитываются.
Существуют 4 основных метода расчета концентрации анализируемого вещества по хроматографическим данным.
1. Метод нормирования (метод внутренней нормализации). Применение данного метода основано на предположении, что на хроматограмме зарегистрированы все вещества, входящие в состав анализируемой смеси, и что доля площади (высоты) каждого пика от суммы площадей (высот) всех пиков соответствует содержанию вещества в массовых процентах. Процентное содержание вещества в анализируемой смеси рассчитывается путём определения площади соответствующего пика как процентной части общей площади всех пиков, за исключением пиков, соответствующих растворителям или реактивам, подвижной фазе или матрице образца. Содержание каждого вещества в смеси в процентах может быть вычислено по формуле:
Если чувствительность детектора различна по отношению к каждому из веществ, то вводят поправочные коэффициенты ki. Относительный коэффициент отклика детектора, обычно называемый фактором отклика, обозначает чувствительность детектора для данного вещества относительно стандартного вещества. Поправочный коэффициент – это число, обратное фактору отклика.

Поправочные коэффициенты рассчитывают относительно основного вещества анализируемой смеси или другого стандартного вещества по формуле:
 где
где
Сi и С0 – концентрация i-го вещества и стандартного вещества соответственно;
Si и S0 – площадь (высота) пика i-го вещества и стандартного вещества соответственно.
Данные коэффициенты могут не учитываться в случае, если они находятся в пределах диапазона 0,8 – 1,2.
При использовании поправочных коэффициентов выражение для расчета количественного содержания приобретает вид:

При проведении испытания на примеси методом нормализации или методом внешнего стандарта с использованием разведения раствора испытуемого образца в качестве раствора сравнения учитывают все указанные в нормативной документации поправочные коэффициенты, значение которых выходит за пределы диапазона 0,8 – 1,2.
2. Метод внешнего стандарта. Концентрацию испытуемого вещества определяют путём сравнения сигнала (пика), полученного на хроматограммах испытуемого раствора, и сигнала (пика), полученного на хроматограммах раствора стандартного образца.
Концентрацию определяемого вещества в испытуемом растворе рассчитывают по формуле:

S и S0 – средние значения площадей (высот) пиков на хроматограммах испытуемого и стандартного растворов соответственно;
С и С0 – концентрации определяемого и стандартного растворов соответственно.
Количественное определение содержания примесей методом внешнего стандарта предпочтительнее проводить с использованием стандартных растворов примесей с концентрациями, близкими к их ожидаемым концентрациям в испытуемом растворе.
В качестве раствора стандартного образца для количественного определения примесей возможно использование раствора основного вещества. В этом случае разведение подбирается таким образом, чтобы концентрация основного соединения в растворе стандартного образца по отношению к его концентрации в испытуемом растворе была близка к ожидаемой концентрации примесей в испытуемом растворе. В этом случае следует учесть факторы отклика примесей по отношению к основному веществу, если их значения выходят за рамки 0,8 — 1,2.
Частным случаем метода внешнего стандарта является метод калибровочной кривой, в ходе которого определяют взаимосвязь между измеренным или обработанным сигналом (у) и количеством (концентрацией, массой и т.д.) определяемого вещества (х) и рассчитывают уравнение калибровочной функции. Результаты испытания рассчитывают из измеренного или обработанного сигнала с помощью обратной функции.
3. Метод внутреннего стандарта. Концентрацию определяемого вещества определяют путём сравнения отношения сигналов (площадей или высот пиков), соответствующих определяемому веществу и внутреннему стандарту, на хроматограмме испытуемого раствора и отношения сигналов (площадей или высот пиков), соответствующих определяемому веществу и внутреннему стандарту, на хроматограмме раствора стандартного образца. Метод внутреннего стандарта основан на введении в анализируемую смесь определенного количества стандартного вещества (внутренний стандарт). В испытуемый и стандартный растворы вводят известные количества внутреннего стандарта, хроматографируют растворы и определяют отношения площадей (высот) пиков определяемого вещества к площади (высоте) пика внутреннего стандарта в испытуемом и стандартном растворах.
Концентрацию определяемого вещества (Х) рассчитывают по формуле:

4. Метод стандартных добавок. В качестве внутреннего стандарта выбирается вещество, отсутствующее в испытуемой пробе, не взаимодействующее с определяемым веществом и другими веществами пробы, обладающее достаточной стабильностью, полностью отделяющееся от веществ пробы и не содержащее примесей с временами удерживания, совпадающими с временем удерживания определяемого вещества. Концентрация внутреннего стандарта должна быть близка к концентрации определяемых веществ, а структура и свойства по возможности аналогичны структуре и свойствам определяемых веществ.
Концентрация определяемого вещества определяется путём сравнения сигнала (площади или высоты пика), соответствующего определяемому веществу, на хроматограмме испытуемого раствора, и сигнала (площади или высоты пика) определяемого вещества на хроматограмме испытуемого раствора с известной добавкой определяемого вещества. Метод стандартных добавок основан на введении в анализируемую смесь известного количества определяемого вещества и сравнения сигналов, полученных для испытуемого раствора со стандартной добавкой и без добавки определяемого вещества. При внесении стандартной добавки стараются минимизировать разбавление испытуемого образца, чтобы измерения раствора со стандартной добавкой и без проходили в одинаковых условиях с одинаковым влиянием матрицы. Количество вводимого в стандартной добавке определяемого вещества должно быть соизмеримо с его предполагаемым содержанием в испытуемом образце. После проведения испытания сравнивают полученные значения интенсивности и рассчитывают количественное содержание определяемого вещества Сх по формуле:

Сстд — концентрация стандартной добавки;
Sx — интенсивность сигнала определяемого вещества (площадь или высота пика) для испытуемого раствора без стандартной добавки;
Sстд+х — интенсивность сигнала определяемого вещества (площадь или высота пика) для испытуемого раствора со стандартной добавкой.
При необходимости формулу корректируют с учетом разбавлений испытуемого раствора за счет введения стандартной добавки.
РЕКОМЕНДАЦИИ ПО РАЗМЕТКЕ И ИНТЕГРИРОВАНИЮ ХРОМАТОГРАММ ПРИ ОПРЕДЕЛЕНИИ ПРИМЕСЕЙ
Если не указано иное, в испытаниях на содержание примесей в качестве порога игнорирования (неучитываемый предел, уровень содержания вещества, при котором и при меньшем содержании пик не принимается во внимание) принимают величину 0,05 % от содержания основного вещества.
В случаях, когда при испытании на примеси требуется определение суммарного содержания примесей или количественного определения примеси, при интерпретации хроматограммы важно выбрать подходящие условия интегрирования и значения порога интегрирования [интенсивность сигнала пика соединения (высоты или площади)], при котором (и при меньшем) пик не учитывается системой сбора данных и не участвует в количественных расчетах.
С целью разметки на хроматограмме всех пиков, подлежащих учету, заданный порог интегрирования системы сбора данных не должен превышать половину порога игнорирования.
Интегрирование площади пика любой примеси, пик которой не полностью разделяется с основным пиком, предпочтительно проводить экстраполяцией смещенной базовой линии (методом тангенциальной касательной). Использование других способов интегрирования должно быть специально оговорено в фармакопейной статье.
ОЦЕНКА ПРИГОДНОСТИ ХРОМАТОГРАФИЧЕСКОЙ СИСТЕМЫ
Испытания пригодности системы являются неотъемлемой частью методики и используются для того, чтобы убедиться в надлежащем функционировании хроматографической системы и обеспечить выполнение предъявляемых к ней требований.
При проведении испытаний используемое оборудование должно быть квалифицировано и способно к функционированию надлежащим образом.
Для оценки пригодности системы указывают:
- требования к параметрам, характеризующим форму пика [эффективность хроматографической системы N и фактор асимметрии (фактор симметрии) As];
- требования к разделительной способности (разрешение между пиками RS или отношение пик — долина p/v);
- требования к воспроизводимости (относительное стандартное отклонение, RSD) значений площади или высоты пиков, а также времен удерживания пиков в случае оценки подлинности соединений;
- требования к чувствительности при проведении испытания на примеси [минимально определяемая концентрация (фактический предел количественного определения) примеси]. Оценивается посредством расчета отношения сигнал/шум для раствора соответствующей концентрации.
Если в фармакопейной статье не указано иное, должны выполняться следующие требования и все дополнительные требования, приведенные в нормативном документе:
- в испытаниях на примеси или количественное содержание для пика на хроматограмме растворе стандартного образца, используемого для количественных определений, значение величины фактора асимметрии (фактора симметрии) As должно находиться в пределах от 0,8 до 1,5;
- разрешение между пиками Rs ³ 1,5 (в случае не полностью разделенных пиков вместо разрешения может быть использовано соотношение p/v. При этом требования к минимальному значению соотношения p/v устанавливаются в фармакопейной статье);
- отношение сигнал/шум для пика вещества, полученное для раствора с концентрацией, равной требуемому уровню минимально определяемой концентрации, должно быть не менее 10. Требуемый уровень минимально определяемой концентрации [фактический предел количественного определения (ПКО)] зависит от того, предполагает ли методика вычисление содержания примесей, или только полуколичественную оценку, когда результат представляется в виде «менее Х» или «не более Х», где Х – допустимое содержание примеси. Для методик, предполагающих вычисление содержания примесей, минимальная определяемая концентрация для применяемой хроматографической системы не должна превышать значение порога игнорирования (если не указано иное – 0,05 % относительно концентрации основного вещества в испытуемом растворе). Для полуколичественных методик минимальная определяемая концентрация для применяемой хроматографической системы не должна превышать максимально допустимое содержание примеси. При необходимости оценки содержания нескольких примесей требуемый уровень минимальной определяемой концентрация для применяемой хроматографической системы определяется примесью, нормы содержания которой наиболее строги. Если в фармакопейной статье не указано иное, то раствор вещества минимально определяемой концентрации для оценки чувствительности детектирования можно приготовить растворением стандартного образца вещества в том же растворителе, который используется для приготовления испытуемого раствора, с уровнем концентрации 0,05 % относительно концентрации основного вещества в испытуемом растворе.
Требования к прецизионности системы в условиях повторяемости при количественном определении основного вещества в субстанциях
При количественном определении основного вещества в фармацевтических субстанциях при содержании основного вещества, близком к 100 %, максимально допустимое относительное стандартное отклонение значений интенсивности (площади или высоты) пика основного вещества для заданного количества повторных введений стандартного раствора RSDmax вычисляется по формуле:
Если в фармакопейной статье не указано иное, то значение RSD не должно превышать величин RSDmax, приведенных в таблице. Данное требование не применяется при испытаниях на примеси.

Таблица. Максимально допустимое относительное стандартное отклонение RSDmax в зависимости от верхнего предела содержания основного вещества и числа отдельных введений проб
| В, % | Число отдельных введений проб (вколов) | |||
| 3 | 4 | 5 | 6 | |
| Максимально допустимое относительное стандартное отклонение RSDmax | ||||
| 2,0 | 0,41 | 0,59 | 0,73 | 0,85 |
| 2,5 | 0,52 | 0,74 | 0,92 | 1,06 |
| 3,0 | 0,62 | 0,89 | 1,10 | 1,27 |
Соответствие критериям пригодности системы должно поддерживаться на протяжении всего испытания. В зависимости от различных факторов, например, частоты использования методики и опыта работы с хроматографической системой, аналитик выбирает соответствующую схему проверки, подтверждающую соответствие этим требованиям.
В планарной хроматографии при проведении испытаний, регламентирующих наличие посторонних примесей/родственных соединений, должно быть предусмотрено определение предела обнаружения определяемых примесей и разделительной способности хроматографической системы.
Для подтверждения пригодности системы возможно одновременное использование двух тестов: «Подтверждение разделительной способности» и «Подтверждение чувствительности».
Тест «Подтверждение разделительной способности» проводят путём нанесения на пластинку и последующего хроматографирования специального стандартного раствора, содержащего два или более веществ с известными значениями Rf. После хроматографирования эти вещества должны разделиться, образуя зоны адсорбции, значения Rf которых равны заданным.
Тест «Подтверждение чувствительности» позволяет оценить чувствительность проявления. Он заключается в нанесении определённого количества стандартного вещества, хроматографируемого в условиях предыдущего теста и проявляемого тем же реагентом. Зона адсорбции стандартного вещества должно чётко обнаруживаться.
В каждом случае природа стандартных веществ, состав стандартного раствора, наносимые количества, приблизительные значения Rf устанавливают в фармакопейных статьях.
При необходимости для достижения требуемых критериев пригодности хроматографической системы проводится корректировка хроматографических условий.
КОРРЕКТИРОВКА УСЛОВИЙ ХРОМАТОГРАФИРОВАНИЯ
Далее приводятся пределы возможных изменений различных параметров хроматографической методики, которые могут быть внесены в нее для соответствия требованиям пригодности системы без принципиального изменения методик. Корректировка условий градиентного элюирования является более критичной, чем изократических условий, так как может вызвать сдвиг пиков на другую стадию градиента, и, таким образом, привести к некорректной идентификации пиков, наложению пиков или сдвигов, при которых элюирование интересующих соединений происходит после указанного времени регистрации хроматограммы. При внесении изменений, отличных от приведенных ниже, требуется проведение повторной валидации методики.
Проверка пригодности системы должна быть включена в методики для подтверждения получения разделения, требуемого для надлежащего проведения испытания. Тем не менее, поскольку неподвижные фазы описаны только в общем виде, и существует огромное количество доступных коммерческих фаз, отличающихся по хроматографическому поведению, некоторая корректировка условий хроматографирования может потребоваться для выполнения требований пригодности системы. В методиках с использованием обращенно-фазовой жидкостной хроматографии корректировки различных параметров не всегда приводят к удовлетворительному разделению. В этом случае может возникнуть необходимость замены одной колонки на другую, такого же типа, обеспечивающую желаемое хроматографическое поведение.
Если в фармакопейной статье не указано иное, допустимы следующие изменения параметров хроматографической системы.
Тонкослойная и бумажная хроматография
Состав подвижной фазы: количество растворителя, содержание которого в смеси наименьшее, может изменяться в пределах ± 30 % (относительное содержание) или ± 2 % (абсолютное содержание) в зависимости от того, какая из величин будет больше. Абсолютное содержание других веществ не может быть изменено более чем на 10 %.
Пример 1: для вещества, содержание которого составляет 10 % подвижной фазы, изменение относительного содержания на 30 % приведет к изменению его абсолютного содержания до 7 – 13 %, тогда как изменение на 2 % по абсолютному содержанию приведет к изменению абсолютного содержания до 8 – 12 %. Допустимое изменение по относительному содержанию больше, чем допустимое изменение по абсолютному содержанию. Таким образом, допускается изменение содержания 7 – 13%.
Пример 2: для вещества, содержание которого составляет 5 % подвижной фазы, изменение относительного содержания на 30 % приведет к изменению его абсолютного содержания до 3,5 – 6,5 %, тогда как изменение на 2 % по абсолютному содержанию приведет к изменению его абсолютного содержания в подвижной фазе до 3 – 7 %. В этом примере допустимое изменение по абсолютному содержанию больше, чем допустимое изменение по относительному содержанию, и допустимым диапазоном содержания является 3 – 7 %.
pH среды водного компонента подвижной фазы: ± 0,2 pH, если иное не указано в нормативном документе, или ± 1,0 pH в случаях испытания неионизируемых веществ.
Концентрация солей в буферном веществе подвижной фазы: ± 10 %.
Наносимый объём. При использовании пластинок с малым размером частиц сорбента (2 — 10 мкм) наносимый объём может быть изменен на 10 –20 % от заявленного объёма.
Жидкостная хроматография: изократическое элюирование
Состав подвижной фазы: количество растворителя, содержание которого в смеси наименьшее, может изменяться в пределах ± 30 % (относительное содержание) или ± 2 % (абсолютное содержание) в зависимости от того, какая из величин будет больше. Абсолютное содержание других веществ не может быть изменено более чем на 10 %.
pH среды водного компонента подвижной фазы: ± 0,2 pH, если иное не указано в нормативном документе, или ± 1,0 pH в случаях испытания неионизируемых веществ.
Концентрация солей в буферном веществе подвижной фазы: ± 10 %.
Скорость потока подвижной фазы: ± 50 %, большая степень изменений допустима при одновременном изменении размеров колонки. При необходимости одновременного изменения размеров колонки и скорости потока сначала рассчитывается номинальная скорость потока для колонки, размеры которой отличаются от приведенной в нормативном документе, а затем допускается корректировка полученного значения скорости потока на ± 50 %.
Параметры колонки
Неподвижная фаза:
- замена типа неподвижной фазы недопустима (например, недопустима замена фазы С18 на фазу С8);
- размер частиц: максимальное уменьшение размера частиц 50 %, увеличение не допускается.
Размеры колонки
Длина колонки: ± 70 %,
внутренний диаметр колонки: ± 25 %.
При изменении размеров колонки скорость потока пересчитывают, используя следующее уравнение:

- F1 — скорость потока, указанная в нормативном документе, мл/мин;
- F2 — скорректированная скорость потока, мл/мин;
- l1 — длина колонки, указанная в нормативном документе, мм;
- l2 — длина используемой колонки, мм;
- d1 — внутренний диаметр колонки, указанный в нормативном документе, мм;
- d2 — внутренний диаметр используемой колонки, мм.
Температура: ± 10 % от указанной рабочей температуры, если не указано иначе.
Длина волны детектора. Изменения не допускаются.
Вводимый объём пробы. Может быть уменьшен при условии, что чувствительность фактически применяемой хроматографической системы (минимально определяемая концентрация) и прецизионность системы в условиях повторяемости для определяемых соединений остаются удовлетворительными.
Жидкостная хроматография: градиентное элюирование
Изменение хроматографических условий для систем с градиентом требует большей осторожности, чем для изократических.
Состав подвижной фазы/профиль градиентного элюирования: незначительные изменения состава подвижной фазы и системы градиента являются приемлемыми при условии, что:
- выполнены требования пригодности системы;
- основной пик элюируется в пределах ± 15 % от обозначенного времени удерживания;
- элюирующая способность конечного состава подвижной фазы не менее предписанного состава.
Если при использовании градиентного элюирования не может быть достигнуто соответствие требованиям пригодности системы, рекомендуется оценить объем задержки градиента или сменить используемую колонку.
Объем задержки градиента. Конфигурация используемого оборудования может значительно изменить разрешение, указанные абсолютные и относительные времена удерживания. Это может произойти из-за отличия объема задержки градиента используемой системы от объема задержки градиента системы, с помощью которой проводилась валидация методики. В нормативном документе часто перед началом программы градиента включена изократическая стадия, чтобы можно было адаптировать систему к временным точкам градиента с учетом разницы объема задержки у системы, использованной для разработки методики, и у фактически использующейся системы. Решение о необходимости адаптации длительности изократической стадии для данного аналитического оборудования принимается пользователем. Если объем задержки, использованный при разработке методики, приведен в нормативном документе, то интервалы времени (t), указанные в таблице градиента, можно заменить адаптированными интервалами времени (tc), рассчитанными с помощью следующего уравнения:

D – объем задержки, мл;
D0 – объем задержки, использованный при разработке методики, мл;
F – скорость потока, мл/мин.
Изократическая стадия, введенная с этой целью, может быть исключена, если имеются данные валидации по применению методики без этой стадии.
pH среды водного компонента подвижной фазы. Изменения не допускаются.
Концентрация солей в буферном веществе подвижной фазы. Изменения не допускаются.
Скорость потока. Изменения допустимы при изменении размеров колонки.
Параметры колонки
Неподвижная фаза:
- замена типа неподвижной фазы недопустима (например, недопустима замена С18 на С8);
- размер частиц: изменения не допускаются;
Размеры колонки
- длина колонки: ± 70 %;
- внутренний диаметр колонки: ± 25 %;
При изменении размеров колонки скорость потока пересчитывают, используя следующее уравнение:

- F1 — скорость потока, указанная в нормативном документе, мл/мин;
- F2 — скорректированная скорость потока, мл/мин;
- l1 — длина колонки, указанная в нормативном документе, мм;
- l2 — длина используемой колонки, мм;
- d1— внутренний диаметр колонки, указанный в нормативном документе мм;
- d2 — внутренний диаметр используемой колонки, мм.
Температура. ± 5 % от указанной рабочей температуры, если не указано иначе.
Длина волны детектора. Изменения не допускаются.
Вводимый объём пробы. Может быть уменьшен при условии, что детектирование (предел количественного определения) и сходимость отклика (RSD площадей или высот) для пика (пиков) определяемых соединений остаются удовлетворительными.
Газовая хроматография
Параметры колонки
Неподвижная фаза:
- размер частиц: максимальное уменьшение на 50 %, увеличение не допускается (набивные колонки);
- толщина плёнки: от минус 50 % до плюс 100 % (капиллярные колонки).
Размеры колонки
- Длина колонки: ± 70 %;
- Внутренний диаметр колонки: ± 50 %.
Скорость потока: ± 50 %.
Температура: ± 10 %.
Вводимый объём пробы. Может быть уменьшен при условии, что чувствительность фактически применяемой хроматографической системы и прецизионность системы в условиях повторяемости для определяемых соединений остаются удовлетворительными.
Сверхкритическая флюидная хроматография
Состав подвижной фазы. Для набивных колонок количество растворителя, содержание которого в смеси наименьшее, может изменяться в пределах ± 30 % (относительное содержание) или ± 2 % (абсолютное содержание) в зависимости от того, какая из величин будет больше; для систем с капиллярной колонкой изменения не допускаются.
Длина волны детектора. Изменения не допускаются.
Параметры колонки
Неподвижная фаза:
- размер частиц: максимальное уменьшение на 50 %, увеличение не допускается (набивные колонки).
Размер колонки
- Длина колонки: ± 70 %;
- Внутренний диаметр колонки: ± 25 % (набивные колонки), ± 50 % (капиллярные колонки).
Скорость потока: ± 50 %.
Температура: ± 5 %, если температура указана в нормативном документе.
Вводимый объём пробы. Может быть уменьшен при условии, что чувствительность фактически применяемой хроматографической системы и прецизионность системы в условиях повторяемости для определяемых соединений остаются удовлетворительными.
There are many methods to separate and identify different compounds. Chromatography is the science that allows you to separate the components of a mixture. The Rf (sometimes informally written as “rf value”) is part of that.
There are many different kinds of chromatography. Here, the focus is the simplest kind of chromatography: paper chromatography or thin layer chromatography (TLC). Both use the same general procedure, which is described below.
What Does Chromatography Involve?
All types of chromatography have a stationary phase and a mobile phase. The stationary phase, as you might be able to guess from the name, does not move. In paper chromatography, the stationary phase is the paper itself.
To begin the separation of compounds, the substance to be analyzed is dotted onto the bottom edge of the piece of paper. Then the paper is placed in a beaker containing the mobile phase, which is a solvent. The solvent should be under the spot where the substance for analysis is dotted. It should not submerge the analyte.
The liquid moves up the paper by capillary action. If the analyte or part of the analyte is attracted to the mobile phase, it will move with it. If it has a stronger attraction for the stationary phase, it will not move.
After the solvent has moved up the stationary phase to some extent, you have a chromatogram. This chromatogram tells you something about the separation of different components in a mixture or tells you about what substance you have. How?
Separating and Identifying Components Using a Chromatogram
If the conditions for developing a chromatogram are the same, then the distance a particular substance moves should change. This distance is measured by the retention factor formula or Rf.
The retention factor formula is:
This value should be same for a substance when the chromatogram is developed in the same way.
This is useful when you have a mixture of substances.
For example, say you have four samples: Three are standards, and one is mixture. Your job is to identify which components are in the mixture.
What are standards? Standards are a pure sample of a substance. This can be run on the chromatogram as a comparison.
The mixture can be run, after which you can determine how far different components in it run.
Say Standard #1 has a strong affinity for water (the mobile phase). This means that it will travel up the paper with the mobile phase. Standard #2 on the other hand has a strong affinity for paper. This means that it will stay with the stationary phase. Lastly, Standard #3 has an affinity for both water and paper. You predict it will move an intermediate amount.
You find that the solvent moves 5.7 cm while Standard #1 moves 0.5 cm, Standard #2 moves 4.8 cm, and Standard #3 moves 2.9 cm. What are the Rf values for each standard?
Now, you know the Rf values for the standards. Next, on another piece of paper you place a dot of the mixture and allow the chromatogram to develop by placing it in water.
Once removed, you find that there are two dots that separated from the initial dot you placed. This likely means that there are only two components in your mixture. Next, you need to figure out which of the standards these two components correspond to. To do this, you measure the Rf values.
The solvent moved 4.5 cm. One component (called component 1) moved 2.2 cm, and the other moved 3.9 cm (called component 2). To see which standard these components correspond to you will need to use the retention factor formula to calculate the Rf values.
Given these Rf values, it is likely that component 1 corresponds to Standard #3 since the Rf values are very similar. Component 2 is likely Standard #2.
-
Хроматографическая подвижность веществ
-
Общие теоретические закономерности
-
Основной характеристикой хроматографической
подвижности вещества, используемой в
ТСХ, является величина Rf,
характеризующая подвижность вещества
относительно подвижности элюентав
данных условиях проведения анализа.
Определяется эта величина (уравнение
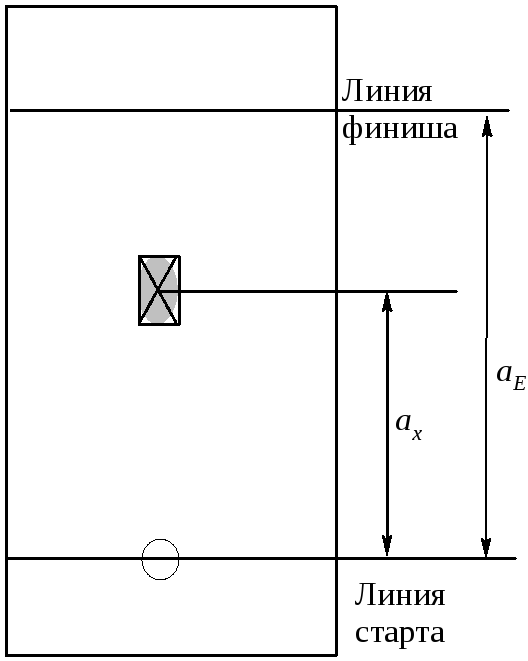
1 .1), как отношение расстояния, пройденного
пятном вещества от линии старта (ax)
к расстоянию между линией старта и
линией финиша (aE). Метод
определения этих величин представлен
на рисунке 3 .1
|
|
( |
1.1 |
) |
|
Рисунок |
3.1 |
– |
Если величина aEпроизвольно
задается условиями проведения анализа,
то величинаaxзависит от
большого количества факторов, значения
которых в большинстве случаев
контролировать сложно, поэтому для
надежной характеристики подвижности
вещества, воспроизводимой от эксперимента
к эксперименту вне зависимости от
условий, лаборатории и времени проведения
анализа следует пользоваться величиной,
характеризующей подвижность изучаемого
вещества относительно подвижности
стандартного вещества в этих условиях
–RS(уравнение 1 .2).
|
|
( |
1.2 |
) |
где Rf(X) иRf(S)
– хроматографическая подвижность
изучаемого и стандартного веществ,
соответственно.
К сожалению, до настоящего времени нет
общепринятого набора стандартов,
позволяющего “привязать” большинство
органических веществ.
Для описания способности вещества
адсорбироваться на неподвижной фазе в
хроматографии часто используетсявеличинаk’–коэффициент удержания– отношение количества вещества в
подвижной фазе к его количеству в
неподвижной, рассчитываемый в ТСХ по
формуле 1 .3
|
|
( |
1.3 |
) |
Однако, гораздо удобнее использовать
величину, обратную k’, которую часто
обозначают как Rm(уравнение
1 .4):
|
|
( |
1.4 |
) |
Предпочтительность такого описания
связана с тем, что величина Rmсимбатна величинеRf(Rmизменяется в том же направлении, что иRf). Еще одним достоинством
этой величины является то, что она в
отличии отRfаддитивна
по струкутрным фрагментам веществаи позволяет априорно приблизительно
оценивать хроматографические свойства
вещества в ряду однотипных.
Следует иметь в виду, что сказанное
выше имеет смысл только в том случае,
если вещество наносится на пластинку
в минимальных количества – его должно
быть не больше, чем может быть растворено
в том количестве элеюнта, которое
находится в объеме “над пятном” и так
мало, чтобы на сорбенте “в пятне”
степень заполнения центров адсорбции
данным веществом стремилась к 0 (была
как можно меньше). Критерием выбора
количества наносимого вещества для
этих целей должен служить предел
обнаружения.
Другими словами эти (и последующие)
соображения справедливы, только тогда,
когда элюция проводится в условиях
ненасыщенности сорбента сорбатом,
и, следовательно, величинаRf
численно равна степени десорбции
вещества с сорбента в элюент.
Если кроме равновесия
![]()
одновременно имеет место ассоциация
вещества в супрамолекулярные комплексы
![]()
или его кристаллизация
![]() ,
,
то наблюдаемое значение Rfбудет отличаться от “истинного”, т.е.
будет характеризовать не свойства
вещества, а особенности процесса.
Исходя из основных законов адсорбции
и расматривая хроматографический
процесс как дискретный (непрерывный
поток элюента можно с определенной
долей надежности описать как набор
большого количества циклов – бесконечно
быстрый скачок элюента на расстояние
одной “зоны” и установление в течение
определенного интервала времени
равновесия между подвижной и неподвижной
фазами), можно вывести уравнение 1 .5
(его вывод являтся несложным, но
громоздким, в связи с чем здесь мы его
пропустим; при выводе используется
допущение о том, что вся поверхность
сорбента однородна1и полностью насыщена элюентом),
описывающегоRfвещества
как функцию от способности вещества и
элюента к адорбции на сорбенте (XиS,
соответственно), состояние поверхности
которого можно описать эмпирическим
безразмерным параметром.
|
|
( |
1.5 |
) |
Зная Rfвещества на сорбенте
с параметромв
одном элюенте (![]() ),
),
можно рассчитать его способность к
адсорбцииX,
а затем –Rfв любом другом
элюенте (![]() )
)
на том же сорбенте (=const). Для многих растворителей
значенияSизвестны (см. ниже – раздел 4.1.3).
При желании можно вывести свой собственный
ряд элюотропных параметров растворителей
(‘S).
Для этого выбирают некоторую группу
веществ и некоторый набор растворителей.
Для произвольного из выбранных веществ
полагаютX
= 1, аполагают
равной 1 (вообще говоря, можно задать
любые действительные числа). Тогда,
получив из результатов хроматографического
эксперимента величинуRf,
можно рассчитать величину‘S
для использованного растворителя.
Для остальных отобранных веществ
значенияXрассчитывают из их величинRf,
полученных на том же сорбенте (= 1) в том же растворителе. Далее, проводя
элюцию этого набора веществ на том же
сорбенте с использованием в качестве
элюентов остальных растворителей можно
рассчитать параметры‘Sэтих растворителей.
Иллюстрацией эффективности такого
подхода могут служить результаты
изучения подвижности 2,7-диалкоксифлуоренонов
на пластинках Silufol в малополярных
элюентах.
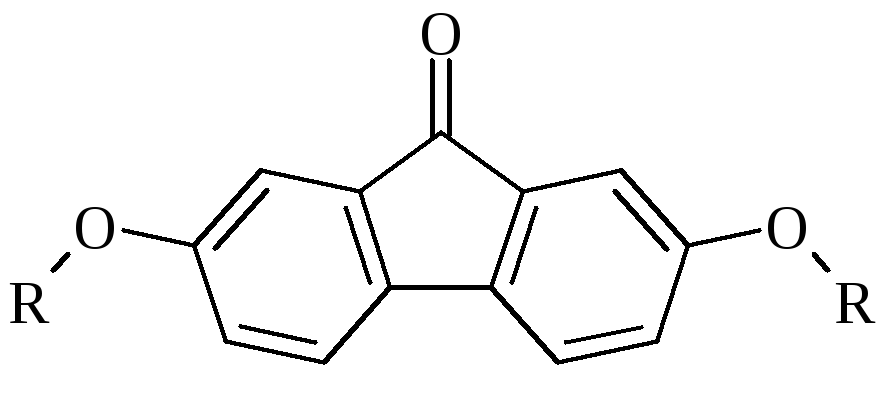 R
R
= CnH2n+1; n = 1, 2, 3, 4, 5, 6, 7, 8, 9, 11,
12.
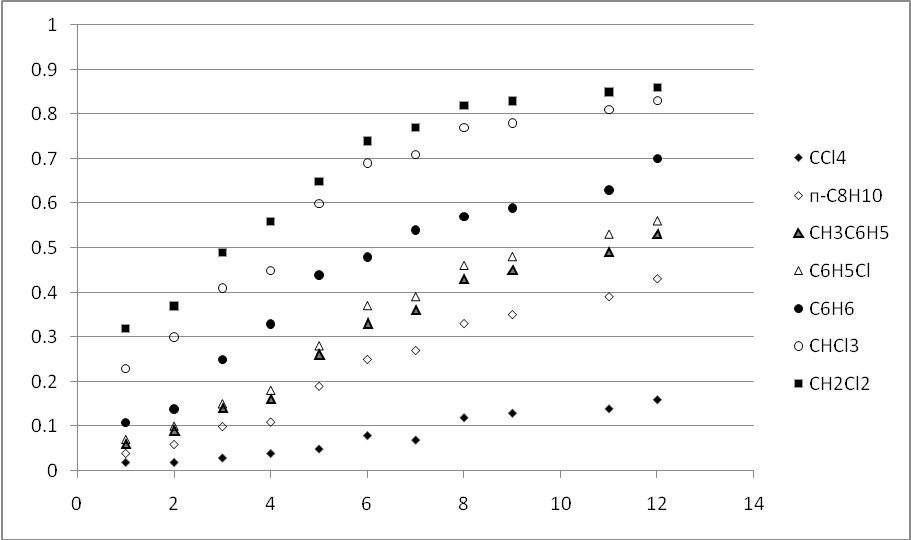
Для этих веществ были получены значения
Rfв четыреххлористом
углероде, хлороформе, хлористом метилене,
бензоле, ксилоле, хлорбензоле,и смесях этих растворителей. Полученные
значения приведены отображены на рисунке
3 .2.
|
Рисунок |
3.2 |
– |
Silufol UV-254 в различных элюентах. На линию
старта пластинок наносили по (8–10) 10-9моль (2–5g)
вещества в 5l раствора
в четыреххлористом углероде.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Как рассчитать значения Rf для TLC
Автор:
Louise Ward
Дата создания:
10 Февраль 2021
Дата обновления:
16 Май 2023

Содержание
- Что такое TLC
- Как рассчитать значения Rf для TLC
Значение Rf представляет собой коэффициент удерживания, используемый при идентификации органических соединений в смеси. Значение Rf рассчитывается путем измерения относительного расстояния, пройденного конкретным органическим соединением относительно подвижной фазы. Он рассчитывается как в бумажной хроматографии, так и в ТСХ.
Тонкослойная хроматография (ТСХ) представляет собой метод разделения, включающий разделение двух или более органических соединений путем их распределения между твердой и жидкой фазами. Стационарная (твердая) фаза представляет собой полярное вещество в ТСХ, тогда как подвижная (жидкая) фаза представляет собой один или комбинацию растворителей. Абсорбент покрыт тонким слоем на предметном стекле. Органические соединения отделяются путем манипулирования подвижной фазой.
Ключевые области покрыты
1. Что такое TLC
– определение, принцип, использование
2. Как рассчитать значения Rf для TLC
– Расчет значения Rf
Ключевые термины: подвижная фаза, мобильность, Rf Value, стационарная фаза, разделение, тонкослойная хроматография (TLC)

ТСХ – это метод хроматографии, отвечающий за разделение органических соединений на смеси на основе их относительной подвижности. Он использует твердую стационарную фазу, состоящую из полярного абсорбента, и жидкую подвижную фазу, состоящую из одного или смеси органических растворителей. Полярным абсорбентом могут быть либо частицы тонко измельченного оксида алюминия, либо частицы диоксида кремния. Разделение черных чернил с помощью ТСХ показано на Рисунок 1.

Рисунок 1: TLC черных чернил
Органические соединения и подвижная фаза движутся через стационарную фазу благодаря капиллярному действию. Дифференциальная подвижность органических соединений достигается за счет относительного сродства соединений к стационарной фазе и подвижной фазе. Соединения с более высоким сродством к стационарной фазе движутся медленно, так как стационарная фаза удерживает органические соединения. Поскольку в ТСХ стационарная фаза является полярной, полярные соединения движутся медленно. Однако соединения с более высоким сродством к подвижной фазе движутся быстрее через стационарную фазу. Подвижная фаза является неполярной, и неполярные органические соединения, которые имеют меньшее сродство к стационарной фазе, быстрее перемещаются через стационарную фазу. Отдельные соединения могут быть визуализированы в виде пятен после разделения.
TLC используется в:
- Определение количества соединения в смеси
- Проверка состава смеси
- Определение подходящих условий для колоночной хроматографии
- Анализ фракций, полученных из колоночной хроматографии
Как рассчитать значения Rf для TLC
Значение Rf является относительным расстоянием, пройденным конкретным соединением относительно подвижной фазы. Это можно рассчитать по следующему уравнению.
Рисунок 2: Расчет значения Rf
Rf = расстояние, пройденное соединением (a) / расстояние, пройденное фронтом растворителя (b)
Значение Rf также называется отношением к фронту. При определенных условиях стационарной фазы, подвижной фазы и температуры значение Rf конкретного органического соединения является постоянным значением. Однако разные органические соединения с одинаковой полярностью могут иметь одинаковые значения Rf. Следовательно, другие свойства органических соединений, такие как цвет на пластине ТСХ, могут быть использованы во время идентификации соединения.
Заключение
ТСХ – это метод хроматографии, используемый для разделения органических соединений на основе их полярности. Дифференциальная полярность этих соединений обеспечивает дифференциальную подвижность через стационарную фазу ТСХ. Значение Rf представляет собой относительную подвижность конкретного органического соединения по отношению к подвижной фазе. Он рассчитывается путем измерения относительных расстояний, пройденных органическими соединениями.
Ссылка:
1. «Тонкослойная хроматография (ТСХ): принцип и методика».Owlcation29 декабря 2015 г.,
Skip to content
Rf values: Definition, Calculation and Explanation
Retardation or retention factor (Rf) value is the ratio of distance traveled by the analyte to that of the solvent front on a chromatogram.
![]()
The chromatographic techniques in which the analytes are added to the stationary phases show a difference in the movement of analytes with mobile solvents (phases). This difference is due to the relative affinities of analytes with stationary and mobile solvents. The more the relative affinity with a stationary phase an analyte has, the more it will stay in place, and the lesser the Rf value will get and vice versa.
Rf value is the characteristic identification value for analytes at given temperatures. It means that compounds can be analyzed and identified based on their Rf values. It is, however, not the case when a new compound is discovered.
Calculation of Rf values
A chromatogram is first to be developed with a suitable solvent (mobile phase), depending upon the nature of analytes, and the stationary phases. The developed chromatogram is then dried and the positions (migration values) are measured for analytes and the solvent front.

The Rf (retardation/retention factor) values can be calculated by using the given procedure using the above experiment.
A prepared sample solution (A+B) is applied on the chromatogram paper and run through a mobile phase. Analyte (A) and (B) separate out because of different affinities with mobile phase (solvent). The relative measurements are taken for the analytes, the solvent front, and the point where the mixture (A+B) was applied.
For the analyte (A)
Rf = Distance moved by analyte (A) / Distance moved by solvent front
Rf = 2.9 / 4.0
Rf = 0.725
For the analyte (B)
Rf = Distance moved by analyte (B) / Distance moved by solvent front
Rf = 1.3 / 4.0
Rf = 0.325
So, the Rf values for the analytes (A) and (B) are 0.725 and 0.325.
Factors affecting Rf values
There are several factors that affect the Rf values of a particular analyte:
- Stationary phase
- Concentration of stationary phase
- Mobile phase
- Concentration of mobile phase
- Temperature
Same substances (analytes) have different Rf values if the nature of stationary and mobile phases is changed, i.e. the affinity factors are changed. Similar is the case when the concentration of these phases is changed. Temperature affects the rate of mobile action of solvent and also the solubilities of analytes in the solvent.
Rf values are independent of the concentration of analytes whatsoever.
Related Topics
- Calibration and its curves
- Relative vs. standard deviation
- Solvent extraction
Concepts Berg
How to calculate Rf?
Retention/retardation factor (Rf) can be calculated by the relative migration values of solute (analyte) and the solvent front.
Rf = Migration of analyte / Migration of solvent front
The calculation of the Rf value is basically the calculation of relative affinities of a solute with the stationary and mobile phases.
How to choose the solvent system?
The solvent system for a particular analyte separation via chromatography is chosen based on the polarity of solute (analyte), the stationary phases, and the temperature. The solvent system is actually the mobile phase which must relatively be either more or less polar than the stationary phase, which becomes the reason for the displacement of the analyte from the application point to a higher level in column chromatography.
A pure solvent can be used with particular polarity for a chromatographic procedure. Although if a series of continuously increasing or decreasing polarities is required, a mixture of two or more solvents may be used with a continuous manner of increase in the concentration of one solvent and decrease of other.
How to interpret the TLC?
Interpretation of the thin layer chromatography (TLC) is totally based on the Rf values. The Rf values evaluate the relative polarity, the relative affinities with stationary and mobile phases, the relative molecular weights, and even the concentration of analytes if sophisticated environments are used.
Why do we need the Rf value?
Rf values in chromatography are the basic requirement of the whole experiment. These values tell us whether the analyte (solute) is more affinitive with stationary or the mobile phase. Rf values evaluate the polarity, relative masses, and relative solubilities with stationary and mobile phases, etc.
How to find the Rf values of aspirin in specific solvents?
Rf values are found by chromatographic experiments. Aspirin is a polar molecule and will be more soluble in a polar solvent. As the general rule of solubility ‘like dissolves like’ suggests. The Rf values are always different for different stationary and mobile phases used. So, an aspirin solution is to be run against a polar solvent (mobile phase) when the stationary phase is less polar than the mobile one. This will leave aspirin at a specific height giving its Rf value between 0 and 1.
What are the units for Rf values?
Rf values are unitless as the ratio of similar quantities is always just a number. The solute migration value is compared by the solvent front migration value to calculate the Rf value which gives just a ratio number.
What are the advantages of calculating an Rf value in chromatography?
Chromatography is extremely useful only if there is an account for Rf values. These values give the necessary information required, for which the chromatography is basically performed. Rf values evaluate the polarities, the relative affinities with stationary and mobile phases, the relative molecular weights, and even the identification of analytes (solutes) are done by the Rf values unless there is no record for that particular solute in those conditions.
Which one is better in TLC, the higher or the lower Rf value?
The basic property of Rf values is to determine the relative properties of components of a mixture among each other and with the stationary and mobile phases. It doesn’t actually matter if their values are higher or lower as long as these are far apart from one another.
Rf values must not be either 0 or 1 because both of these conditions are not useful in the analytical procedure. An Rf value of zero means that the complete displacement (migration) of the analyte with the mobile phase (solvent), whereas the value one refers to no displacement at all.
Is chromatography paper polar?
In column chromatography, the chromatography paper is made of a cellulose network (like a normal paper) having capillary action to move solvent over it. The compound acting as the stationary phase is the adsorbed water on cellulose fibers which is a polar compound. So, the chromatographic paper in column chromatography is polar. This is also the reason why it is called liquid-liquid chromatography.
In thin layer chromatography, the TLC paper or sheet is having alumina (AL2O3) or silica (SiO2) pasted over glass or metal sheet, which are polar so a TLC paper is also polar.
What is the principle behind chromatography?
The main principle of chromatography is the relative solubilities of a single analyte in different solvents.
Retardation/retention factor (Rf) values drive the whole procedure because it is the Rf values that determine the relative polarities/solubilities of components in a mixture (solutes) based on the polarities of stationary and mobile solvents.
The principle of chromatography can thus be, the relative affinities of analytes (solutes) with stationary and mobile phases.
The principle of relative affinities is further described by the adsorption or dissolution of solute on stationary solvent or phase and then leaving the stationary solvent for a relatively higher solubility in mobile solvent/phase.
In chromatography, if solvent reaches the edge of the paper so how can we determine the Rf value?
In chromatographic techniques, the solvent front is usually not let to reach the end of the paper. The development of chromatogram is stopped before that happens and is placed to dry for further evaluation. But if somehow, the solvent front reaches the edge of paper during chromatography, the value of solvent front migration is taken to be the length of the paper.
What is a good Rf value?
Rf values are the retardation values in a specific environment for a particular analyte (solute). These values aren’t good or bad. These are just a ratio of two migration distances occurring simultaneously, the analyte and the solvent front. It would be better for calculation and identification purposes if the Rf values of compounds of a mixture are not too close.
Although, if the Rf values of compounds in a mixture are too close, these would not be of much use.
Why is Rf less than 1?
Rf values are always less than one because they are the ratios of migration distances of solutes (analytes) and solvent fronts. As a general rule, the solvent front always travels more than that of solute, because the solutes have to have some attractive properties with stationary phases. It means that the denominator being higher in value, Rf values will always be between 0 and 1.
What is the significance Rf value?
Rf values are fairly significant because the outcome of most of the chromatographic procedures, especially TLC and column chromatography rest on these Rf values. It is Rf values that provide the relative properties of analytes like polarities, molecular weights, affinities with particular solvents, and even the identification of analytes (solutes) can only be achieved through the Rf values library.
Related Articles
Page load link
Go to Top
 report this ad
report this ad
