КИСЛОТНОСТЬ И ОСНОВНОСТЬ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Кислотность и основность – важнейшие понятия, определяющие многие фундаментальные физико-химические свойства и биологическую активность органических соединений.
Теории кислот и оснований:
— теория электролитической диссоциации (Аррениус);
— протолитическая теория (Бренстед-Лоури, 1923 г.);
— теория Льюиса (1925 г.).
Протолитическая теории Бренстеда
Кислота – это вещество, способное отдавать протон. Основание – это вещество, способное присоединять протон. Кислота + основание = сопряженная пара

Кислоты
Атом, с которым связан протон, – центр кислотности. Центрами кислотности могут быть атомы C, O, N, S: Соответственно – CH-, OH-, NH- и SH-кислоты.
Основания
Центрами основности являются атомы, имеющие неподеленную пару электронов (n-электроны): N, O, S. Соответственно различаются аммониевые, оксониевые и тиониевые основания (n-основания). Это могут быть нейтральные молекулы или анионы.
Существуют также π-основания – соединения с кратными связями или сопряженной системой π-связей. Они присоединяют протон, образуя сопряженные кислоты – π-комплексы.
Количественная оценка кислотности
Сила кислоты – это степень сдвига вправо следующего равновесия:
![]()
Количественной мерой кислотности являются:
Ка – константа кислотности
рКа = -lgKa – показатель кислотности
сила кислоты↑ → Ка↑ → рКа↓
Количественная оценка основности

Сила основания – это степень сдвига вправо следующего равновесия:
Из этих соотношений следует, что для сопряженной пары мерой основности основания В может быть кислотность сопряженной кислоты ВН+: рКВ = 14 – рКВН+
Сила основания↑ → КВ↑ → рКВ↓ → рКВН+↑
Качественная оценка кислотности
Сила кислоты определяется устойчивостью сопряженного основания (аниона).
Чем стабильнее анион, тем сильнее кислота.
Устойчивость аниона, в свою очередь, зависит от следующих факторов:
— свойства атома в центре кислотности – его электроотрицательность и поляризуемость
— степень делокализации (-)-заряда в анионе в результате сопряжения;
— действие окружающих заместителей;
— способность аниона к сольватации.
Факторы, определяющие кислотность
1. Свойства атома в центре кислотности
а) электроотрицательность
С увеличением электроотрицательности атома Х полярность связи Х-Н увеличивается, прочность её уменьшается, облегчается отрыв протона. В результате кислотность увеличивается. ЭО↑ → Кислотность↑
б) поляризуемость
С увеличением радиуса атома Х увеличивается длина связи и её поляризуемость, связь легче разрывается, кислотность увеличивается: Радиус атома↑ → Кислотность↑
Ряд увеличения кислотности: CH < NH < OH < SH
2. Делокализация заряда по сопряженной системе
Делокализация заряда по сопряженной системе увеличивает стабильность аниона и соответственно увеличивает кислотность.

2. Влияние заместителей
а) электроноакцепторы
Электроноакцепторы способствуют делокализации заряда в анионе, увеличивают его устойчивость и, следовательно, увеличивают кислотность.
б) электронодоноры
Электронодоноры препятствуют делокализации заряда в анионе, уменьшают его устойчивость и, следовательно, уменьшают кислотность.
3. Эффект сольватации

Сольватация образующего аниона способствует его устойчивости и увеличивает кислотность. (!) Чем меньше размер иона и чем больше в нем локализован заряд, тем легче он сольватируется.
Качественная оценка основности
Наиболее сильные основания – анионы: HO—, RO—, NH—2, SH—
 Нейтральные молекулы в реакциях с кислотами образуют ониевые соли:
Нейтральные молекулы в реакциях с кислотами образуют ониевые соли:

Выделяются три типа органических оснований:
Сила основания определяется стабильностью образующегося катиона, а также доступностью
неподеленной пары электронов для присоединения протона. На силу основания влияют те же факторы, что и на силу кислоты, но направление действие их противоположно.
Амины – наиболее сильные основания. Они образуют с кислотами устойчивые соли растворимые в воде. Это свойство аминов широко применяется для их выделения и очистки, а также для получения растворимых лекарственных форм.
Ряд уменьшения основности: R-NH-R > R-O-R > R-S-R
— основность оксониевых оснований уменьшается за счет большей ЭО кислорода;
— тиониевые основания более слабые, чем оксониевые потому, что больший радиус серы увеличивает длину связи S-H в катионе, делает её менее прочной и тем самым снижает устойчивость катиона.
На силу оснований большое влияние оказывают заместители у центра основности:
а) электроноакцепторы
Электроноакцепторы увеличивают (+)-заряд на катионе, уменьшают его устойчивость и, следовательно, уменьшают основность.

б) электронодоноры
Электронодоноры уменьшают (+)-заряд на катионе, увеличивают его устойчивость и, следовательно, увеличивают основность.

Включение неподеленной электронной пары в сопряженную систему понижает её доступность для присоединения протона и снижает основность:

Амиды:
Из-за такого сопряжения основность амидов по сравнению с аминами резко снижена, в водных растворах амиды не протонируются (их основность ниже, чем у воды):
Теория кислот и оснований Льюиса
Кислота – акцептор электронов; Основание – донор электронов
При взаимодействии кислот и оснований образуются донорно-акцепторные комплексы:

You have no doubt heard of acids and can probably name a few just from reading food labels: Citric acid. Acetic acid. At the same time, you know that at least some acids can be harmful if you so much as handle them, so different acids clearly have different properties, including different strengths.
Bases are everywhere in the world, too, although they seem to get less publicity for some reason. Like acids, bases can be damaging to biological and other materials. You’ve encountered a strong base in the form of household laundry bleach (NaClO, or sodium hypochlorite).
Acids and bases are complementary in almost every way, and one can even be used to “neutralize” the other, as with taking oral antacid tablets to combat stomach acid. Part of this is in the nomenclature; when acids actually behave like acids, they become bases, and ditto for the behavior of bases. Understanding conjugate acids and bases is essential to mastering chemical reactions.
History of Acid-Base Chemistry
As far back as the mid-1600s, Robert Boyle, who seemed to be involved in just about every chemistry experiment in those days, figured out that certain solutions had properties such as the ability to damage immersed substances or change their colors, and that these effects could be prevented or negated by the addition of alkali compounds, which today are known to be basic.
In 1923, Johannes Brønsted and Thomas Lowry formally defined acids and bases in terms of the transfer of hydrogen ions (H+).
Brønsted-Lowry Acids
The conjugate base of an acid is the compound remaining after a hydrogen ion is donated by the acid, and the conjugate acid of an base is the compound remaining after a hydrogen ion is accepted by the base.
A Brønsted-Lowry acid is therefore simply a molecule that can donate a hydrogen ion (which is a positively charged atom) to another molecule; the remnant of that acid is called its conjugate base. For example, when hydrochloric acid donates a proton, the chloride ion left behind is the conjugate base:
HCl → H++ Cl−
Sometimes, an acid will be positively charged before donating its hydrogen ion, rather than neutral as in the instance of HCl. This can be observed with the ammonium ion donating a proton to become the conjugate base ammonia:
NH4+ → H++ NH3
H2PO4− : Acid or Base?
So far, you have seen examples of compounds with formulas that make it obvious whether the molecule functions as an acid or as a base (or, for that matter, as neither). If you see an ion with no hydrogen atoms included, such as Cl−, you know that it cannot be an acid, since it has no protons, but that it could be a base, since it is an anion with a charge of −1 and “eager” to take on a proton.
But what about compounds with multiple hydrogen atoms available for exchange? In the right environment, a compound that functions as a base in the presence of a strong enough acid can also act as an acid in the presence of a strong enough base. (Think of bases as “hydrogen-ion-pullers.” Such a compound is called amphoteric or amphiprotic.
A classic example is the dihydrogen phosphate ion H2PO4− . In the presence of the strong acid HBr, this molecule readily accepts the hydrogen ion from the acid to become phosphoric acid (H3PO4). Yet in the presence of basic hydroxide (OH−) ions, dihydrogen phosphate instead donates a proton to become monohydrogen phosphate (HPO42−).
-
The conjugate base of H2PO4−
is therefore HPO42−, and the conjugate acid of
H2PO4− is H3PO4.
По протолитической, или протонной теории кислота – это соединение, частица которого может отдать протон (Н + ) другой частице – основанию. Согласно этой теории кислоты и основания – это вещества, теряющие и приобретающие протоны и называемые протолитами. Передача протона от кислоты к основанию называется протолизом. Кислота–донор протонов,а основание–акцептор Н + .
В результате отдачи протона сама кислота превращается в сопряженное ей основание. Основание – акцептор Н + – превращается в сопряженную ему кислоту.
НА – кислота, А – – сопряженное ей основание; В – основание, НВ + – сопряженная ему кислота.
Таким образом, в равновесной протолитической реакции присутствуют две сопряженные пары «кислота / основание»: НА /А – и НВ + / В.
Кислоты и основания, или протолиты, могут быть нейтральными (молекулы), катионными или анионными:
Протонодонорная способность вещества – это его кислотность; протоноакцепторная способность – это основность. Эти свойства определяются сродством к протону, т.е. величиной энтальпии реакции присоединения протона.
При кислотно-основном взаимодействии роль основания играет тот партнер, у которого протонное сродство выше.
1) NH3 – более сильный акцептор протона и более сильное основание, чем H2O, а NH4 + , наоборот, более слабый донор протона и более слабая кислота, чем H3O + ;
2) OH – – более сильное основание, чем F – , а H2O – более слабая кислота, чем HF.
Количественной характеристикой состояния протолитического равновесия обратимой реакции между кислотой HA и основанием (растворителем) HL
Чем больше значение Kк, тем более сильной кислотой является протолит HA в данном растворителе HL.
Количественной характеристикой состояния протолитического равновесия в реакции между основанием B и кислотой (растворителем) HL
Kо = [HB + ] . [L – ] / [B] = Const = f(T). (16)
Чем больше значение Kо, тем более сильным основанием является протолит B в данном растворителе HL.
Пример 2.1.
где HCl – кислота, а Н2О – основание.
Кислоты – молекулы или ионы, способные в данной реакции отдавать катион водорода
Кислота – донор протонов
Основания – молекулы или ионы, способные в данной реакции присоединять протоны (акцепторы протонов)
Основания – акцепторы протонов.
С каждой кислотой сопряжено основание, в которое она переходит, теряя протоны.
Каждой кислоте соответствует свое основание и каждому основанию – своя кислота. Эту пару сопряженных веществ называют кислотно-основной или протолитической парой. Каждый отдельный компонент этой пары называется протолитом. Кислота и основание одной протолитической пары называются сопряженными протолитами (сопряженными кислотой и основанием).
Чем сильнее кислота, тем слабее сопряжённое с ней основание.
нейтр. к-та основание сопряжённая к-та сопряжённое основание
2) Катионные кислоты, представляющие собой положительные ионы, например NH4 + , H3O + :
3) Анионные кислоты, представляющие собой отрицательные ионы, например HSO4 – , H2PO4 – , HPO4 2- и др.
2) Анионные основания, представляющие собой отрицательные ионы, например Cl – , CH3COO – , OH – :
3) Катионные основания, представляющие собой положительные ионы, например H2N-NH3 +
Связь между константой кислотности и константой основности в сопряженной протолитической паре.
К равновесию, которое устанавливается в растворе слабого электролита между молекулами и ионами, можно применить законы химического равновесия и записать выражение константы равновесия. Например, для электролитической диссоциации (протолиза) уксусной кислоты, протекающей под действием молекул воды,
Здесь в числителе дроби стоят равновесные концентрации ионов – продуктов диссоциации, а в знаменателе – равновесная концентрация недиссоциированных молекул.
Константа протолитического равновесия, определяющая полноту протекания протолиза слабой кислоты при данной температуре, называется константой кислотности.
существует константа основности, определяющая полноту протекания протолиза слабого основания при данной температуре
Величины Ка и Кb для сопряженной кислотно-основной пары связаны также простым соотношением.
Амфолиты.
Амфолиты — амфотерные электролиты, т. е. вещества, молекулы которых содержат одновременно и кислотные, и основные группы, и поэтому в водных растворах диссоциируют и как кислоты с отщеплением водородных ионов Н + , и как основания с отщеплением гидроксильных ионов ОН – .
К амфолитам относятся биологически важные вещества: аминокислоты , пептиды , белки и др. Кислотные свойства этих веществ обусловлены наличием в них карбоксильных групп СООН, а основные свойства — содержанием аминогрупп NH2. К амфолитам также относится вода.
Теория Льюиса.
Кислоты Льюиса – это молекулы или ионы, имеющие вакантные электронные орбитали, вследствие чего они могут акцептировать электронную пару. (пример – протон водорода, катионы металлов, оксиды некоторых неметаллов (SO3, SiO2), ряд солей (AlCl3 и др.))
Основания Льюиса – это молекулы и ионы, имеющие пару электронов, являющиеся донорами электронных пар. Примеры: все анионы, аммиак, вода, спирты, галогены и др.
Взаимодействие между кислотой и основанием заключается в образовании донорно-акцепторной связи между реагирующими частицами.
Принцип жестких и мягких кислот (ЖМКО).
Основан на теории Льюиса.
Жёсткие кислоты – это акцепторы электронной пары, обладающие малым размером, значительным положительным зарядом, большой электроотрицательностью и низкой поляризуемостью.
Примеры: протоны водорода, катиона лития, магния, калия, натрия, алюминия, хрома. Молекулы BF3, AlCl3 и др.
Жёсткие основания – донорные частицы с высокой электроотрицательностью, низкой поляризуемостью. Соединения прочно удерживают электроны.
Примеры: гидроксид-ион, фторид-ион, хлорид-ион, нитрат-ион, NH3, R-NH2, H2O, спирты, эфиры.
Мягкие кислоты – кислоты Льюиса, содержащие акцепторные атомы с малым положительным зарядом, большим размером, низкой электроотрицательностью, высокой поляризуемостью.
Мягкие основания – основания Льюиса, содержащие донорные частицы с низкой электроотрицательностью, высокой поляризуемостью.
Слабо удерживают валентные электроны.
Суть принципа ЖМКО: жёсткие кислоты реагируют с жёсткими основаниями, мягкие кислоты – с мягкими основаниями.
Билет 18. Автопротолиз воды. Константа автопротолиза воды. Водородный показатель (pH) как количественная мера активной кислотности и основности. Определение активной концентрации ионов водорода.
Автопротолиз воды.
Автопротолиз – обратимый процесс образования равного числа катионов и анионов из незаряженных молекул жидкого индивидуального вещества за счет передачи протона от одной молекулы к другой.
Для воды характерна протолитическая амфотерность. Реакция самоионизации (автопротолиза) воды, в ходе которой протон от одной молекулы воды (кислоты) переходит к другой молекуле воды (основанию) описывается уравнением:
1.Протолитическая теория кислот и оснований Бренстеда-Лоури (теория сопряжённых пар)(1923 г.). Согласно этой теории:
· Кислоты – это молекулы или ионы, способные отдавать протон, то есть доноры протонов.
· Основания – это молекулы или ионы, способные присоединять протон, то есть акцепторы протонов.
Согласно протолитической теории, кислоты, основания и амфолиты являются протолитами, а процесс перехода протона от кислоты к основания называется протолизом. Подобный подход позволяет оценивать кислотно-основные свойства ионов. Например, карбонат-ион может присоединить протон и, следовательно, является основанием, а гидрокарбонат-ион может отдать протон, и значит, является кислотой: (CO3-)основание + (H+) ⇆ (HCO3-)кислота.
Кислоты Бренстеда-Лоури делят на 3 типа:
· Нейтральные кислоты: HCl, H3PO4, HNO3, HClO
· Катионные кислоты: NH4+, H3O+
· Анионные кислоты: H2PO4-, HCO3-, HSO4-
На такие же три группы делятся основания:
· Нейтральные основания: NH3, H2O
· Анионные основания: OH-, F-
· Катионные основания: (NH2-NH3)+
Согласно протолитической теории, отдавая протон, кислота превращается в частицу, которая называется сопряжённым основанием. Соответственно, основание, присоединяя протон, превращается в сопряжённую кислоту. Кислота и сопряжённое ей основание (или основание с сопряжённой кислотой) образуют сопряжённую кислотно-основную пару, в которой чем сильнее кислота, тем слабее сопряжённое с ней основание и наоборот. Молекулы воды (H2O), аммиака (NH3 жидк.), анионы многоосновных кислот могут быть как донорами, так и акцепторами протонов, т.е. являются веществами-амфолитами.
2. В 1893 г. швейцарским химиком-неоргаником Альфредом Вернером (1866–1919) была сформулирована теория, позволившая понять строение и некоторые свойства комплексных соединений и названная координационной теорией.
Строение комплексных соединений:
Комплексные соединения – это устойчивые химические соединения сложного состава, в которых обязательно имеется хотя бы одна связь, возникшая по донорно-акцепторному механизму.
Согласно теории Вернера центральное положение в комплексных соединениях занимает, как правило, ион металла, который называют центральным ионом, или комплексообразователем.
Комплексообразователь – частица (атом, ион или молекула), координирующая (располагающая) вокруг себя другие ионы или молекулы.
Комплексообразователь обычно имеет положительный заряд, является d-элементом, проявляет амфотерные свойства, имеет координационное число 4 или 6. Вокруг комплексообразователя располагаются (координируются) молекулы или кислотные остатки – лиганды.
Лиганды – частицы (молекулы и ионы), координируемые комплексообразователем и имеющие с ним непосредственно химические связи (например, ионы: Cl–, I–, NO3–, OH–; нейтральные молекулы: NH3, H2O, CO).
Координационное число – это число химических связей, которые комплексообразователь образует с лигандами.
Комплексообразователь и окружающие его лиганды составляют внутреннюю сферу комплекса. Частица, состоящая из комплексообразователя и окружающих лигандов, называется комплексным ионом. При изображении комплексных соединений внутреннюю сферу (комплексный ион) ограничивают квадратными скобками. Остальные составляющие комплексного соединения расположены во внешней сфере.
3.Задача на рН буферной системы.
S(Ag3PO4)=0,0065 г/л Ag3PO4 -> 3Ag+ + PO4-
5. Определить концентрации
K2[Cd(CN)4]2- -> 2K+ + [Cd(CN)4]2-
[Cd(CN)4] -> Cd2+ + 4CN-
ПИ=1,248*10^-14 * 0,05 = 6,25*10^-16
ПИ > Ks – осадок выпадает
1.Теория Льюиса опирается на строение внешних электронных оболочек атомов и позволяет объяснить свойства соединений, не содержащих H+.
Кислотой Льюиса называют вещества, имеющие вакантные орбитали, и способные принимать электронные пары, то есть быть акцептором электронов. К ним относятся H+, Na+, K+, Mg+2, Ca+2, Mn+2, Al+3, Ag+, Cu+, Hg+2 и т.д.
Основание Льюиса – вещества, имеющие не поделённые электронные пары и способные их отдавать для образования химической связи, то есть быть донорами электронов. К ним относятся H2O, OH-, ROH, NH3, RNH2, ROR, Cl-, F- и т.д. Отличительной способностью кислот и оснований является их взаимная нейтрализация путем образования ковалентной связи.
Согласно теории Льюиса, различают жёсткие и мягкие кислоты и основания. В соответствии с принципом ЖМКО (жёстких и мягких кислот и оснований) жёсткие кислоты преимущественно реагируют с жёсткими основаниями, а мягкие кислоты – с мягкими основаниями.
Классификация кислот и оснований в рамках принципа ЖМКО:
· Жёсткие кислоты (H+, Li+, Na+, K+, Mg2+, Ca2+, Al3+, Cr3+, Fe3+, BF3, B(OR)3, AlR3, AlCl3, SO3, BF3, RCO+, CO2, RSO2+);
· Жёсткие основания (OH-, RO-, F-, Cl-, RCOO-, NO3-, NH3, RNH2, H2O, ROH, SO42-, CO32-, R2O, NR2-, NH2-);
· Промежуточные кислоты (Cu2+, Fe2+, Zn2+, SO2, R3C+, C6H5+, NO+);
· Промежуточные основания (Br-, C6H5NH2, NO2-, C5H5N);
· Мягкие кислоты (Ag+, Cu+, Hg2+, RS+, I+, Br+, Pb2+, BH3, карбены);
· Мягкие основания (RS-, RSH, I-, H-, R3C-, алкены, C6H6, R3P, (RO)3P).
2. Классификация КС по заряду иона.
Комплексные соединения – это устойчивые химические соединения сложного состава, в которых обязательно имеется хотя бы одна связь, возникшая по донорно-акцепторному механизму.
По характеру заряда комплексного иона комплексные соединения бывают:
─ катионные (положительный заряд внутренней сферы), например, [Cu(NH3)4]Cl2 (комплексный ион [Cu(NH3)4]
─ анионные (отрицательный заряд внутренней сферы), например,
К2[Cd(NO2)4] (комплексный ион [Cd(NO2)4]-2);
─ нейтральные (представлены только одной внутренней сферой, которая является электронейтральной), например, Cr(H2O)3Cl3]o.
Буферными системами (буферами) называют растворы, обладающие свойством сохранять постоянство концентрации водородных ионов как при добавлении небольшого количества кислот или щелочей, так и при разбавлении. По составу различают:
· Кислотные буферы, состоящие из слабой кислоты и её соли, образованной сильным основанием; Например, ацетатная буферная система (CH3COOH+ СН3СООNa ), гидрокарбонатная буферная система (H2CO3 +NaHCO3 ).
· Основные буферы, состоящие из слабого основания и её соли, образованной сильной кислотой; Например, аммиачная буферная система (NH3⋅H2O + NH4Cl).
· Солевые буферы, состоящие из двух кислых или кислой и средней солей. Например, карбонатная буферная система (NaHCO3+Na2CO3), фосфатная буферная система (КН2PO4 + К2НPO4).
Буферное действие сохраняется в определенном интервале значений рН. Рабочий участок буферной системы, т.е. способность противодействовать изменению рН при добавлении кислот и щелочей, имеет протяженность одну единицу рН с каждой стороны. Вне этого интервала буферная емкость быстро падает до 0. Интервал рН = рК ± 1 называется зоной буферного действия.
2. Номенклатура КС.
Комплексные соединения – это устойчивые химические соединения сложного состава, в которых обязательно имеется хотя бы одна связь, возникшая по донорно-акцепторному механизму.
Наибольшее распространение имеет номенклатура, рекомендованная IUPAC. Название комплексного аниона начинается с обозначения состава внутренней сферы: число лигандов обозначается греческими числительными: 2–ди, 3–три, 4–тетра, 5–пента, 6–гекса и т.д., далее следуют названия лигандов, к которым прибавляют соединительную гласную «о»: Cl– – хлоро-, CN– – циано-, OH– – гидроксо- и т.п. Если у комплексообразователя переменная степень окисления, то в скобках римскими цифрами указывают его степень окисления, а его название с суффиксом -ат: Zn – цинкат, Fe – феррат(III), Au – аурат(III). Последним называют катион внешней сферы в родительном падеже.
K3[Fe(CN)6] – гексацианоферрат(III) калия,
K4[Fe(CN)6] – гексацианоферрат(II) калия,
K2[Zn(OH)4] – тетрагидроксоцинкат калия.
Названия соединений, содержащих комплексный катион, строятся из названий анионов внешней среды, после которых указывается число лигандов, дается латинское название лиганда (молекула аммиака NH3 – аммин, молекула воды H2O – аква от латинского названия воды) и русское название элемента-комплексообразователя; римской цифрой в скобках указывается степень окисления элемента-комплексообразователя, если она переменная. Например:
[Cu(NH3)4]SO4 – сульфат тетраамминмеди(II),
[Al(H2O)6]Cl3 – хлорид гексаакваалюминия.
1.Кислотные буферы. Уравнение Г-Г.
Кислотные. Состоят из слабой кислоты и соли этой кислоты. Например, ацетатная буферная система (CH3COOH+ СН3СООNa), гидрокарбонатная буферная система (H2CO3 +NaHCO3).
Если к буферному раствору попеременно добавлять в небольших количествах сильную кислоту или щелочь, то его буферное действие сможет сохраняться более длительное время, т.к. в результате протекающих реакций буферная система будет периодически восстанавливать свой первоначальный количественный и качественный состав.
Механизм буферного действия рассмотреть на примере фосфатного буфера: NaH2PO4 + Na2HPO4.
Добавленная к нему сильная кислота провзаимодействует с солевой компонентой системы и заместится на эквивалентное количество компоненты, играющей роль слабой кислоты.
Na2HPO4 + HCl = NaH2PO4 + NaCl – молекулярное уравнение
2Na+ + HPO42– + H+ + Cl– = 2Na+ + H2PO4– + Cl– – полное ионное уравнение
HPO42– + H+ = H2PO4– – сокращенное ионное уравнение
рН буферной системы рассчитывают по уравнению Гендерсона-Гассельбаха:
· pH = pKa + lg[соль]/[кислота] – для кислотного буфера;
· pH = 14 – pKb – lg[соль]/[основание] – для основного буфера.
2. Хелатные и внутрикомплексные соединения
Циклические комплексные соединения, в образовании которых принимают участие полидентатные лиганды, называются хелатами. Хелатные комплексы отличаются повышенной прочностью. Это относится как к термической прочности, так и к устойчивости внутренней сферы в водных растворах.
Если помимо координационной связи, полидентатный лиганд связан с комплексообразователем еще и ковалентной связью, то образуется дополнительный хелатный цикл. Такие соединения называются внутрикомплексными. Соединения, которые образуют внутрикомплексные соединения с ионами металлов, называются комплексоны. К внутрикомплексным соединениям относятся многие биоло-гические комплексы, например, хлорофилл (комплексообразователь – Mg2+), гемоглобин (Fe2+), витамин В12 (Co3+) и многие другие.
1.Механизм действия основного буфера.
Если к буферному раствору попеременно добавлять в небольших количествах сильную кислоту или щелочь, то его буферное действие сможет сохраняться более длительное время, т.к. в результате протекающих реакций буферная система будет периодически восстанавливать свой первоначальный количественный и качественный состав.
Добавленная к нему сильная кислота провзаимодействует со слабым основанием и заместится на эквивалентное количество солевой компоненты буфера:
NH3 × H2О + HCl = NH4Cl + H2О – молекулярное уравнение
NH3 × H2О + H+ + Cl– = NH4+ + Cl– + H2О – полное ионное уравнение
NH3 × H2О + H+ = NH4++ H2О – сокращенное ионное уравнение
Щелочь вступит в реакцию с солью буферной системы и вместо нее образуется эквивалентное количество слабого основания:
NH4Cl + NaOH = NH3 × H2О + NaCl – молекулярное уравнение
NH4+ + Cl– + Na+ + OH– = NH3 × H2О + Na+ + Cl– – полное ионное уравнение
NH4+ + OH– = NH3 × H2О – сокращенное ионное уравнение
рН буферной системы рассчитывают по уравнению Гендерсона-Гассельбаха:
• pH = pKa + lg[соль]/[кислота] – для кислотного буфера;
• pH = 14 – pKb – lg[соль]/[основание] – для основного буфера.
2. Поведение КС в растворах
Комплексные соединения – это устойчивые химические соединения сложного состава, в которых обязательно имеется хотя бы одна связь, возникшая по донорно-акцепторному механизму.
В водных растворах комплексные соединения диссоциируют на ионы внутренней координационной сферы и ионы внешней сферы по типу сильных электролитов, например:
K3[Fe(CN)6] → 3K+ + [Fe(CN)6]3-
Диссоциация внутренней координационной сферы протекает по типу слабых электролитов и характеризуется константой нестойкости, например:
Kн = [Fe3+] [CN-]6 / [Fe(CN)63-].
Способность буферной системы противодействовать смещению реакции среды измеряется буферной ёмкостью. Буферную ёмкость выражают количеством моль-эквивалентов сильной кислоты или основания, которое следует добавить к одному литру буферного раствора, чтобы сместить на единицу: B = nэ/|pH1-pH2|⋅Vб.р., где:
· B – буферная ёмкость;
· nэ – количество моль-эквивалентов кислоты или щёлочи;
· pH1 – водородный показатель до добавления сильной кислоты или щёлочи;
· pH2 – водородный показатель после добавления сильной кислоты или щёлочи.
· Ацидоз — это сдвиг кислотно-щелочного состояния в связи с положительным балансом водородных ионов, т.е. при накоплении Н-ионов в крови.
· Алкалоз — это сдвиг кислотно-щелочного состояния в связи с отрицательным балансом водородных ионов, т.е. при уменьшении Н-ионов в крови.
2. Хим. Основы хелатотерапии.
Хелатотерапия – это выведение токсичных частиц из организма, основанное на хелатировании их комплексонатами s–элементов.
Препараты, применяемые для выведения инкорпорированных в организме токсичных частиц, называют детоксикантами (Lg). Хелатирование токсичных частиц комплексонатами металлов (Lg) преобразует токсичные ионы металлов (Мт) в нетоксичные (МтLg) связанные формы, подходящие для изоляции и проникновения через мембраны, транспорта и выведения из организма. Они сохраняют в организме хелатообразующий эффект как по лиганду (комплексону), так и по иону металла. Это обеспечивает металлолигандный гомеостаз организма. Поэтому применение комплексонатов в медицине, животноводстве, растениеводстве обеспечивает детоксикацию организма.
Общая кислотность – это концентрация всех катионов водорода (свободных и связанных), имеющихся в растворе при данных условиях.
Активная кислотность (рН) – это показатель, характеризующий активность ионов водорода, который численно равен отрицательному десятичному логарифму их концентрации.
Потенциальная кислотность представляет собой «запас» непродиссоциированных молекул кислоты и может быть вычислена вычитанием из общей кислотности активной.
2. Сущность комплексонометрии.
Комплексонометрия – метод количественного титриметрического анализа, основанный на образовании комплексных соединений ионов металлов с комплексонами.
Способы фиксирования конечной точки титрования. При комплексонометрическом титровании используют металлохромные индикаторы (металлоиндикаторы).
Условия комплексонометрическоro титрования:
1. Реакции комплексообразования должны протекать быстро, количественно и стехиометрично, чтобы вблизи точки эквивалентности определяемые катионы были практически полностью связаны в комплекс. Константа нестойкости образующихся комплексов должна быть малой величиной.
2. Определяемые ионы должны образовывать с металлоиндиктором менее прочные комплексы, чем их комплексы с трилоном Б.
3. Комплексонометрическое титрование следует проводить при определенном значении рН (рН 2I-
С помощью прямого титрования определяют концентрацию восстановителей, непосредственно взаимодействующих с йодом. Обратное титрование заключается в том, что определенный объем раствора добавляют к анализируемому раствору восстановителя, выжидают некоторое время, а затем и титруют избыток не вошедшего в реакцию йода раствором тиосульфата натрия. В качестве индикатора в иодиметрии применяют свежеприготовленный (обычно 1%-ный) раствор крахмала, который окрашивается в Cuний цвет в присутствии даже следовых количеств иода.
Коэффициент активности – это отношение активности вещества в растворе к его концентрации. Он показывает, насколько реальная активность вещества отличается от идеальной.
Коэффициент активности ионов связан связан с ионной силой разбавленного раствора следующим соотношением:
lg f = -0,5Z^2√I, из которого следует, что чем больше ионная сила раствора, тем меньше коэффициент активности его ионов. Если ионные силы двух растворов равны, то коэффициенты активности ионов с одинаковой степенью окисления также равны.
2. Хар-ка хим.связей в КС
В кристаллических комплексных соединениях с заряженными комплексами связь между комплексом и внешнесферными ионами ионная, связи между остальными частицами внешней сферы – межмолекулярные (в том числе и водородные). В молекулярных комплексных соединениях связь между комплексами межмолекулярная.
В большинстве комплексных частиц между центральным атомом и лигандами связи ковалентные. Все они или их часть образованы по донорно-акцепторному механизму (как следствие – с изменением формальных зарядов). В наименее прочных комплексах (например, в аквакомплексах щелочных и щелочноземельных элементов, а также аммония) лиганды удерживаются электростатическим притяжением. Связь в комплексных частицах часто называют донорно-акцепторной или координационной связью.
Теория кислот и оснований Бренстеда-Лоури (теория сопряжённых пар) (1923г.) Согласно этой теории:
· Кислоты – это молекулы или ионы, способные отдавать протон, то есть доноры протонов.
· Основания – это молекулы или ионы, способные присоединять протон, то есть акцепторы протонов.
Согласно протолитической теории, кислоты, основания и амфолиты являются протолитами, а процесс перехода протона от кислоты к основания называется протолизом. Подобный подход позволяет оценивать кислотно-основные свойства ионов. Например, карбонат-ион может присоединить протон и, следовательно, является основанием, а гидрокарбонат-ион может отдать протон, и значит, является кислотой: (CO3-)основание + (H+) ⇆ (HCO3-)кислота Кислоты Бренстеда-Лоури делят на 3 типа:
· Нейтральные кислоты: HCl, H3PO4, HNO3, HClO
· Катионные кислоты: NH4+, H3O+
· Анионные кислоты: H2PO4-, HCO3-, HSO4-
На такие же три группы делятся основания:
· Нейтральные основания: NH3, H2O
· Анионные основания: OH-, F-
· Катионные основания: (NH2-NH3)+
Согласно протолитической теории, отдавая протон, кислота превращается в частицу, которая называется сопряжённым основанием. Соответственно, основание, присоединяя протон, превращается в сопряжённую кислоту. Кислота и сопряжённое ей основание (или основание с сопряжённой кислотой) образуют сопряжённую кислотно-основную пару, в которой чем сильнее кислота, тем слабее сопряжённое с ней основание и наоборот.
2. Иодометрия: сущность заместительного титрования.
Иодометрия – это метод титриметрического анализа, основанный на определении количества йода, которое затрачивается на окисление восстановителей или выделяется при взаимодействии окислителя с раствором иодида калия. I2^0+2e 2I-
Используя заместительное (косвенное) титрование можно определять концентрацию окислителей, взаимодействующих с иодидами с образованием молекулярного йода. Количество выделившегося йода будет эквивалентно количеству определяемого окислителя, а выделившийся йод оттитровывают рабочим раствором тиосульфата натрия. Применяя иодиды в качестве восстановителя, можно провести количественное определение свободного хлора в водных растворах и в хлорной извести CaOCl2.
1.Гет.равн-е. Усл-е обр-я осадка
Гетерогенное равновесие – это количественная характеристика равновесия между раствором и осадком малорастворимого электролита.
Образование осадка может происходить только из пересыщенного раствора. Чтобы выяснить, будет ли происходить осаждение малорастворимого вещества необходимо с учетом разбавления рассчитать произведение концентраций ионов (ПИ) в растворе малорастворимого соединения.
ПК Ks – пересыщенный раствор
Потенциометрия – это совокупность методов физикохимического анализа, основанных на измерении электрохимического
потенциала электрода, функционально связанного с концентрацией
определяемого вещества, называемого потенциалопределяющим.
Таким образом, интенсивноаналитического сигнала в потенциометрии является потенциал электрода..
Электрод, потенциал которого связан с концентрацией
называют индикаторным (ИЭ). Второй электрод – электрод
сравнения (ЭС), в качестве которого обычно используют электроды II
рода, например хлорсеребряный электрод (ХСЭ). ХСЭ представляет
собой серебряную проволоку, покрытую малорастворимой солью
AgCl и помещенную в насыщенный раствор хлорида калия.
Общая форма уравнения Нернста:
1.Гет.равн-е. Растворение осадка
Гетерогенное равновесие – это количественная характеристика равновесия между раствором и осадком малорастворимого электролита.
Осадок малорастворимого электролита может раствориться, если раствор над ним станет ненасыщенным. Следовательно, условием для растворения осадка является соотношение ПИ Ca5(PO4)3 + 3H2O
Потенциометрия – это совокупность методов физикохимического анализа, основанных на измерении электрохимического
потенциала электрода, функционально связанного с концентрацией
определяемого вещества, называемого потенциалопределяющим.
Сущность потенциометрии, а также методов анализа этой области заключается в проведении измерения разности электродвижущей силы двух электродов. Один из них является измерительным, а другой – вспомогательным. К тому же оба эти электрода должны быть помещены в исследуемый раствор. Очень важно отметить, что величина электродвижущей силы сильно зависит от температуры. По этой причине все приборы, которые используются для проведения анализа, в частности рН-метры и ионометры дополнительно комплектуются температурными компенсаторами, которые могут управляться как в ручном режиме, так и в автоматическом.
Потенциометрический метод анализа основан на измерении ЭДС гальванических элементов. Под этими элементами как раз и понимают соединение двух электродов, один из которых индикатор, а другой необходим для сравнения.
1.Сущность метода осадительного титрования.
Метод объединяет титриметрические определения, основанные на реакциях образования осадков малорастворимых соединений. В этих целях пригодны только некоторые реакции, удовлетворяющие определенным условиям. Реакция должна протекать строго по уравнению и без побочных процессов. Образующийся осадок должен быть практически нерастворимым и выпадать достаточно быстро, без образования пересыщенных растворов. К тому же необходимо иметь возможность определять конечную точку титрования с помощью индикатора. Наконец, явления адсорбции (соосаждения) должны быть выражены при титровании настолько слабо, чтобы результат определения не искажался.
Наименования отдельных методов осаждения происходят от названий применяемых титрантов. Метод, использующий раствор нитрата серебра, называют аргентометрией. Этим методом определяют содержание ионов С1- и Вг- в нейтральных или слабощелочных средах.
Тиоцианатометрия основана на применении раствора тиоцианата аммония NH4SCN (или калия KSCN) и служит для определения следов Сl- и Br-, но уже в сильнощелочных и кислых растворах. Используют ее и для определения содержания серебра в рудах или сплавах.
Аргентометрический метод определения галогенов постепенно вытесняется меркурометрическим. В последнем используют раствор нитрата ртути (I) Hg2 (NO3)2.
Метод Мора является одним из аргентометрических методов. Все эти методы основаны на реакции:
Метод Мора является наиболее простым из всех методов аргентометрии и в то же время достаточно точным. Титрантом является раствор нитрата серебра АgNОз 0.1 моль/л.
В основу метода Мора положена реакция взаимодействия ионов серебра с С1- или Вг-.
Индикатором является хромат калия К2СгО4 5 %. Установочным веществом для определения титра раствора является NаС1 или КС1 0.1 моль/л.
Метод Мора позволяет определить количество хлоридов или бромидов. Иодиды этим методом не определяют, так как выпадающий в осадок иодид серебра АgI сильно адсорбирует К2СгО4, поэтому точку эквивалентности определить невозможно.
2. Классификация КС по внешней сфере.
[spoiler title=”источники:”]
http://megalektsii.ru/s4359t7.html
http://topuch.ru/bilet-1-protoliticheskaya-teoriya-kislot-i-osnovanij-brensteda/index.html
[/spoiler]

Елена Шаповалова Владимировна
Эксперт по предмету «Химия»
Задать вопрос автору статьи
Определение кислот и оснований по Бренстеду и Лаури
В $1923$ г. независимо друг от друга Бренстед и Лаури предложили определение кислот и оснований, учитывающих положение о том, что протон в растворе существовать не может. Согласно их определению,
-
кислота — это соединение, частицы которого (ионы или молекулы) в результате протонотранспортной реакции способны отдавать протон другому веществу;
-
основание — это соединение, частицы которого (ионы или молекулы) в результате протонотранспортной реакции принимают протон от другого вещества.
И основание, и кислота во время реакции могут переносить протон, поэтому их называют протолитами. Реакции между протолитами называются протолитическими.
Исходя из этого, кислоты являются протон – донорными веществами, а основания считают протон – акцепторными веществами.
![]()
Сделаем домашку
с вашим ребенком за 380 ₽
Уделите время себе, а мы сделаем всю домашку с вашим ребенком в режиме online
Бесплатное пробное занятие
*количество мест ограничено
- Кислота ↔ Основание + $H^+$
Однако данное соотношение не описывает истинную реакцию (протон $H^+$ в растворе не существует).
В растворе происходит соперничество за протон, в котором участвует и растворитель. Кислоту и основание, связанные за счет обмена протоном, называют сопряженной кислотно — основной парой:
-
Кислота $1$ ↔ сопряженное основание $1$+ $H^+$;
-
Основание $2$ + $H^+$ ↔ сопряженная кислота $2$.
В общем виде реакцию между кислотой и основанием можно записать следующим образом:
- Кислота $1$ + Основание $2$ ↔ сопряженное основание $1$ + сопряженная кислота $2$.
Кислота $1$ будет превращаться в сопряженное с ней основание, и, наоборот, основание $2$ превращается в сопряженную с ним кислоту.
Реакции между кислотами и основаниями Бренстеда
Кислотные взаимодействия характеризуются как реакции между кислотами и основаниями. Если в растворе нет основания — акцептора протона, то вещество как кислота реагировать не может. При этом необходимо присутствие более сильного основания, способного оторвать протон от кислоты. Протон отщепляется от кислоты не самопроизвольно, а под действием основания. Нельзя разделять кислотные и основные свойства, так как существует единое кислотно — основное взаимодействие.
«Кислоты и основания Бренстеда» 👇
Вода является амфотерным растворителем и может выступать как в роли кислоты, так и в роли основания.
Пример 1
-
Вода в роли кислоты, является донором протона:
- $RNH_2 + H_2O ↔ RN{H_3}^+ + OH^-$;
- $HS{O_4}^- + H_3O^+ ↔ H_2SO_4 + H_2O$;
- $CH_3{CO}^{2-} + H_2O ↔ CH_3CO_2H + OH^-$.
-
Вода в роли основания, действует как акцептор протонов:
- $HS{O_4}^- + H_2O ↔ H_3O^+ +{SO_4}^{2-}$;
- $CH_3CO_2H + H_2O ↔ H_3O^+ + CH_3CO^{2-}$.
Ионы могут обнаруживать двойные функции: выступать и как основание и как кислота. Свойства конкретного соединения зависят от свойств, проявляемых конкурирующей сопряженной пары.
Пример 2
Гидрокарбонат — анион $HCO_3$- может проявлять двойственный характер взаимодействия с водой.
-
ведет себя как донор (кислота):
- ${HCO}_{{3^-}(p)} + H_2O(ж) > {H_3O^+}_{(p)} + {{CO_3}^{2-}}_{(p)}$
-
ведет себя как акцептор (основание):
- ${HCO}_{{3^-}(p)} + H_2O(ж) > {H_2CO}_{3 (p)} + {OH^-}_{(p)}$
Определение 1
Вещества – протолиты, способные отдавать и присоединять протоны, называются амфолитами или амфипротонными веществами.
Свойства кислот и оснований противоположны и при взаимодействии они нейтрализуют друг друга, то есть протекает реакция нейтрализации. В результате данной реакции образуется соль (ионное соединение) и вода.
Согласно теории Бренстеда — Лоури, кислоты можно рассматривать как гидратированные ионы металлов. Вода, которая находится во внутренней сфере ионов, выступает донором протонов.
Протолитическая теория описывает многие равновесия. Примеры таких процессов изображены в таблице:
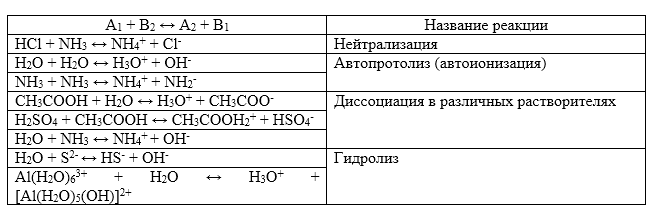
Рисунок 1.
Кислотно – основное титрование – одно из важнейших применений реакции нейтрализации. С его помощью определяют концентрации основания или кислоты в растворе, широко используется в аналитической химии.
При определении концентрации кислоты, пользуются стандартным раствором основания (т.е. с известной концентрацией). И наоборот, при определении концентрации основания пользуются стандартным раствором кислоты.
Количественная характеристика силы кислоты
Кислотно – основные взаимодействия необходимо рассматривать с позиций термодинамики, так как оно является достаточно быстрым и равновесным процессом.
Количественно кислотность вычисляют по отношению к воде. При этом мерой кислотности выступает константа равновесия реакции (константа кислотности) $Ka$:
$A ↔ B + H^+$
Измерить абсолютное значение Кa невозможно, так как данное равновесие возможно при наличии сопряженной пары. Значение константы кислотности можно предопределить относительно выбранного стандарта.
Стандартное равновесие имеет вид:
$H_3O^+ ↔ H_2O + H^+$, $Ka = 1;$
Тогда для любого кислотно — основного процесса:
$A_1 + B_2 ↔ A_2 + B_1$
Константы кислотности кислоты при взаимодействии с водой будут теми же, что и в теории Аррениуса. Для многоосновных кислот первая константа равновесия всегда больше последующих.
Находи статьи и создавай свой список литературы по ГОСТу
Поиск по теме
Как уже было
отмечено выше, основания Бренстеда –
акцепторы протона. Присоединяя протон,
основание переходит в сопряженную
кислоту. Чтобы данное соединение могло
присоединить протон, образовав с ним
ковалентную связь, оно должно иметь
либо свободную пару электронов, либо
π-связь. Например,
![]()
Основание сопряженная сопряжённое Бренстеда кислота. Основание
(имеет пару
электронов)
CH2=CH2
+
H+
= CH3-CH2
+
основание Бренстеда
сопряженная кислота
(содержит π –
связь)
Таким образом,
основаниями Бренстеда могут быть
отрицательно заряженные ионы (самые
сильные основания), молекулы, содержащие
атом с неподелённой парой электронов,
например, амины, спирты, простые эфиры
и самые слабые основания – соединения,
содержащие кратные связи (алкены, арены.)
В соответствии с
этим, все основания Бренстеда делят на
два больших класса: π-основания и
n-основания
(или ониевые). К π-основаниям
относят алкены, алкадиены, арены. Ониевые
основания делят на аммониевые, оксониевые
и сульфониевые. У π-оснований
основным центром , т.е. местом присоединения
протона, являются электроны π-связи.

π-основание
π-комплекс карбокатион
(сопряженная
кислота)
У ониевых оснований
местом присоединения протона являются
соответственно атомы азота, кислорода
и серы.
![]()
первичный
амин- алкиламмоний-
основание
сопряженная кислота

простой
эфир- диалкилоксоний –
основание
сопряженная кислота

тиоэфир-
диалкилсульфоний –
основание
сопряженная кислота

тиоспирт-
алкилсульфоний –
основание
сопряженная кислота
Факторы, влияющие на силу оснований бренстеда
Основность
соединения обычно оценивают по величине
рКа кислоты, сопряжённой с данным
основанием.
основание
+ Н
сопряженная кислота
Чем сильнее кислота,
тем больше её константа диссоциации и
тем меньше её рКа
(pKa
= -lgKa).
Следовательно, чем меньше рКа
сопряжённой кислоты, тем сильнее эта
сопряжённая кислота и тем слабее
основание, из которого она образовалась,
и наоборот, чем больше рКа сопряжённой
кислоты, тем слабее кислота и тем сильнее
основание, из которого эта кислота
получилась в результате присоединения
протона (рКа
сопряжённой с основанием кислоты
обозначают как рКа
ВН![]()
).
1) Факторы, влияющие на силу π-оснований Бренстеда
Алкены, алкадиены
и арены легче присоединяют протон в том
случае, если электронная плотность
π-связей
увеличена. Следовательно, при наличии
электронодонорных
заместителей сила π-оснований
возрастает, а при наличии электроноакцепторных
заместителей – уменьшается.
Так, например,
присоединение протона к этилену,
плотность π-электронов
в котором распределяется симметрично,
протекает труднее, чем реакция
взаимодействия протона с метилпропеном,
где электронная плотность увеличена,
а симметрия её нарушена за счёт
положительного индуктивного эффекта
метильных групп,

кроме того,
присоединение протона приводит к
образованию устойчивого третичного
карбокатиона.
С другой стороны,
присоединение протона к этилену протекает
легче, чем к хлористому винилу, где
π-электроны
двойной связи смещены к сильно-
электроноакцепторному атому хлора, на
котором электронная плотность становится
наибольшей.
Атом углерода, к
которому присоединяется протон, несет
некоторый положительный заряд
![]()
Образовавшийся
карбокатион стабилизирован электронодонорным
эффектом метильного радикала и смещением
р- электронов атома хлора к пустой
орбитали атома углерода.
(Подробное объяснение
такому необычному присоединению протона
к винилхлориду рассмотрено в главе
«Углеводороды»).
2) Факторы,
влияющие на силу ониевых оснований
Бренстеда.
Здесь влияние
природы заместителей прямо противоположно
тому действию, которое оказывают эти
заместители на силу кислот Бренстеда.
а)
Электроотрицательность атомов основного
центра.
Чем больше электроотрицательность
атома основного центра, тем сильнее
электронная плотность смещена к ядру,
и тем труднее данный атом присоединяет
протон. Следовательно, аммониевые
основания должны быть сильнее оксониевых
(Э.О.
азота меньше
Э.О. кислорода).
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
