Гидроксисоединения – это органические вещества, молекулы которых содержат, помимо углеводородной цепи, одну или несколько гидроксильных групп ОН.
Гидроксисоединения делят на спирты и фенолы.
Строение, изомерия и гомологический ряд спиртов
Химические свойства спиртов
Способы получения спиртов

Если гидроксогруппа ОН соединена с бензольным кольцом, то вещество относится к фенолам.
Общая формула предельных нециклических спиртов: CnH2n+2Om, где m ≤ n.
Получение спиртов
1. Щелочной гидролиз галогеналканов
При взаимодействии галогеналканов с водным раствором щелочей образуются спирты. Атом галогена в галогеналкане замещается на гидроксогруппу.
Например, при нагревании хлорметана с водным раствором гидроксида натрия образуется метанол
![]()
Например, глицерин можно получить щелочным гидролизом 1,2,3-трихлорпропана:

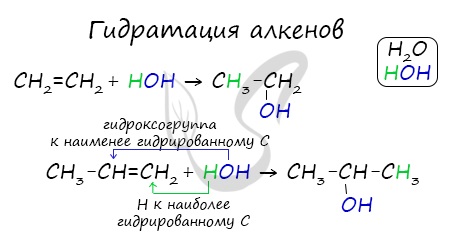
2. Гидратация алкенов
Гидратация (присоединение воды) алкенов протекает в присутствии минеральных кислот. При присоединении воды к алкенам образуются спирты.
Например, при взаимодействии этилена с водой образуется этиловый спирт.
![]()
Гидратация алкенов также протекает по ионному (электрофильному) механизму.
Для несимметричных алкенов реакция идёт преимущественно по правилу Марковникова.
Например, при взаимодействии пропилена с водой образуется преимущественно пропанол-2.

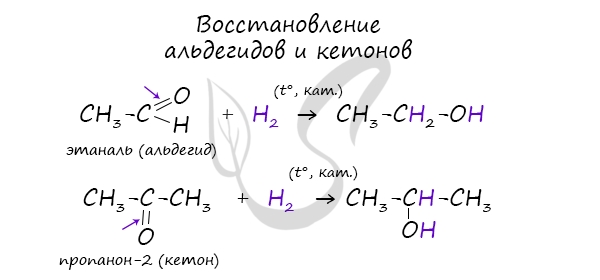
3. Гидрирование карбонильных соединений
Присоединение водорода к альдегидам и кетонам протекает при нагревании в присутствии катализатора. При гидрировании альдегидов образуются первичные спирты, при гидрировании кетонов — вторичные спирты, а из формальдегида образуется метанол.
Например, при гидрировании этаналя образуется этанол

Например: при гидрировании ацетона образуется изопропанол

Например, гидрирование диальдегида – один из способов получения этиленгликоля

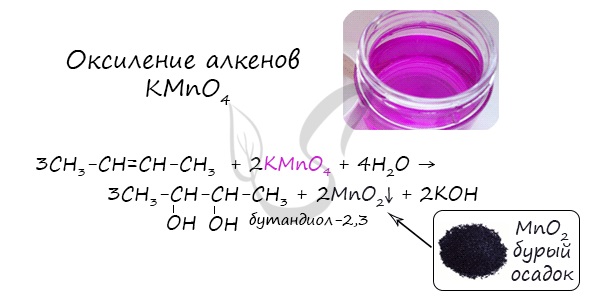
4. Окисление алкенов холодным водным раствором перманганата калия
Алкены реагируют с водным раствором перманганата калия без нагревания. При этом образуются двухатомные спирты (гликоли).

5. Промышленное получение метанола из «синтез-газа»
Каталитический синтез метанола из монооксида углерода и водорода при 300-400°С и давления 500 атм в присутствии смеси оксидов цинка, хрома и др.
Сырьем для синтеза метанола служит «синтез-газ» (смесь CO и H2), обогащенный водородом:
CO + 2H2 ⇄ CH3OH
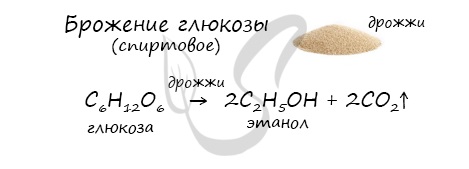
6. Получение этанола спиртовым брожением глюкозы
Для глюкозы характерно ферментативное брожение, то есть распад молекул на части под действием ферментов. Один из вариантов — спиртовое брожение.
![]()
7. Гидролиз жиров – способ получения многоатомных спиртов
Под действием кислоты жиры гидролизуются до глицерина и карбоновых кислот, которых входили в молекулу жира.
Например: при гидролизе тристеарата глицерина образуется глицерин и стеариновая кислота

При щелочном гидролизе жиров образуется глицерин и соли карбоновых кислот, входивших в состав жира.
Например: при щелочном гидролизе тристеарата глицерина образуется глицерин и соль стеариновой кислоты (стеарат)

Спирты – кислородсодержащие органические соединения, функциональной группой которых является гидроксогруппа (OH) у
насыщенного атома углерода.
Спирты также называют алкоголи. Первый член гомологического ряда – метанол – CH3OH.
Общая формула их гомологического ряда – CnH2n+1OH.
Классификация спиртов
По числу OH групп спирты бывают одноатомными (1 группа OH), двухатомными (2 группы OH – гликоли), трехатомными (3 группы
OH – глицерины) и т.д.
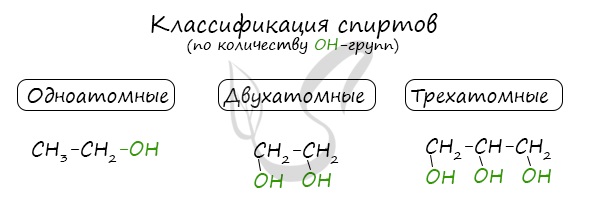
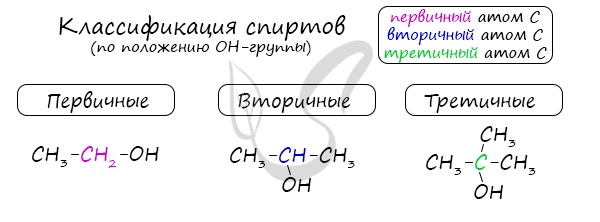
Одноатомные спирты также подразделяются в зависимости от положения OH-группы: первичные (OH-группа у первичного атома углерода),
вторичные (OH-группа у вторичного атома углерода) и третичные (OH-группа у третичного атома углерода).
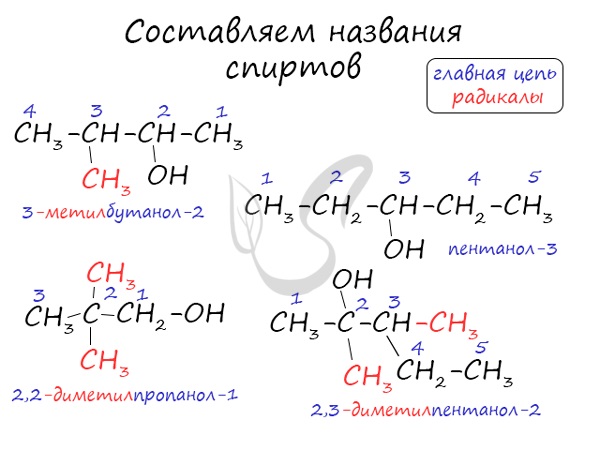
Номенклатура и изомерия спиртов
Названия спиртов формируются путем добавления суффикса “ол” к названию алкана с соответствующим числом атомов углерода: метанол,
этанол, пропанол, бутанол, пентанол и т.д.
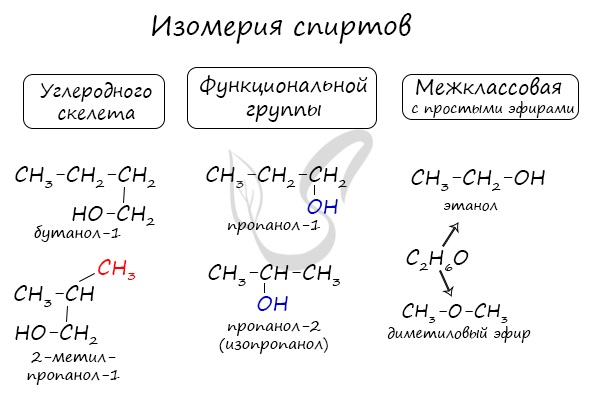
Для спиртов характерна изомерия углеродного скелета (начиная с бутанола), положения функциональной группы и межклассовая изомерия с
простыми эфирами, которых мы также коснемся в данной статье.
Получение спиртов
- Гидролиз галогеналканов водным раствором щелочи
- Гидратация алкенов
- Восстановление карбонильных соединений
- Получение метанола из синтез-газа
- Получение этанола брожением глюкозы
- Окисление алкенов KMnO4 в нейтральной (водной) среде
Помните, что в реакциях галогеналканов со сПИртовым раствором щелочи получаются Пи-связи (π-связи) – алкены, а в реакциях с водным раствором
щелочи образуются спирты.

Присоединения молекулы воды (HOH) протекает по правилу Марковникова. Атом водорода направляется к наиболее гидрированному атому углерода,
а гидроксогруппа идет к соседнему, наименее гидрированному, атому углерода.
В результате восстановления альдегидов и кетонов получаются соответственно первичные и вторичные спирты.
Синтез газом в промышленности называют смесь угарного газа и водорода, которая используется для синтеза различных
химических соединений, в том числе и метанола.
CO + 2H2 → (t,p,кат.) CH3-OH
В ходе брожения глюкозы выделяется углекислый газ и образуется этанол.
В результате такой реакции у атомов углерода, прилежащих к двойной связи, формируются гидроксогруппы – образуется двухатомный спирт (гликоль).
Химические свойства спиртов
Предельные спирты (не содержащие двойных и тройных связей) не вступают в реакции присоединения, это насыщенные кислородсодержащие соединения.
У спиртов проявляются новые свойства, которых мы раньше не касались в органической химии – кислотные.
- Кислотные свойства
- Реакция с галогеноводородами
- Реакции с кислотами
- Дегидратация спиртов
- Диметиловый эфир – CH3-O-CH3
- Метилэтиловый эфир – CH3-O-C2H5
- Диэтиловый эфир – C2H5-O-C2H5
- Окисление спиртов
- Качественная реакция на многоатомные спирты
- Кислотные свойства многоатомных спиртов
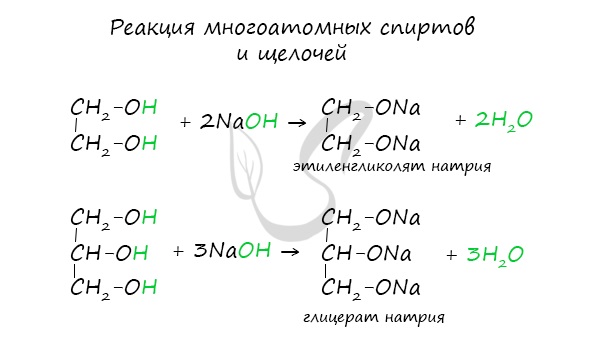
Щелочные металлы (Li, Na, K) способны вытеснять водород из спиртов с образованием солей: метилатов, этилатов, пропилатов и т.д.

Необходимо особо заметить, что реакция с щелочами (NaOH, KOH, LiOH) для предельных одноатомных спиртов невозможна, так как образующиеся
алкоголяты (соли спиртов) сразу же подвергаются гидролизу.
Реакция с галогеноводородами протекают как реакции обмена: атом галогена замещает гидроксогруппу, образуется молекула воды.
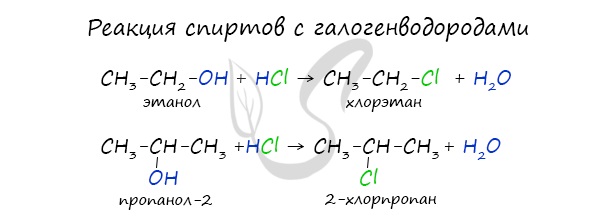
В результате реакций спиртов с кислотами образуются различные эфиры.

Дегидратация спиртов (отщепление воды) идет при повышенной температуре в присутствии серной кислоты (водоотнимающего) компонента.
Возможен межмолекулярный механизм дегидратации (при t < 140°С), в результате которого образуются простые эфиры. При более
высокой температуре (t > 140°С) механизм дегидратации становится внутримолекулярный – образуются алкены.
Названия простых эфиров формируются проще простого – по названию радикалов, входящих в состав эфира. Например:

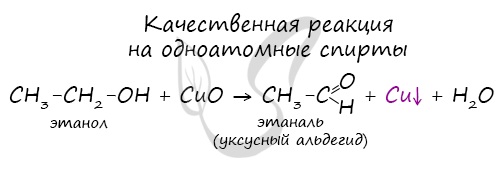
Качественной реакцией на спирты является взаимодействие с оксидом меди II. В ходе такой реакции раствор приобретает характерное фиолетовое
окрашивание.
Замечу, что в обычных условиях третичные спирты окислению не подвергаются. Для них необходимы очень жесткие условия, при
которых углеродный скелет подвергается деструкции.
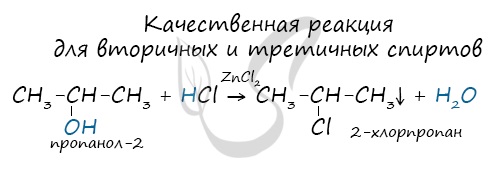
Вторичные и третичные спирты определяются другой качественной реакцией с хлоридом цинка II и соляной кислотой. В результате такой
реакции выпадает маслянистый осадок.
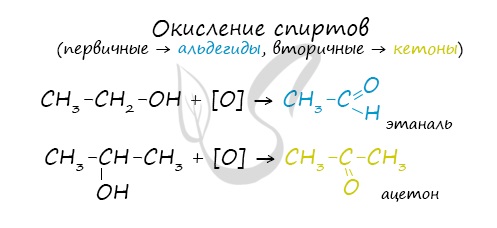
Первичные спирты окисляются до альдегидов, а вторичные – до кетонов. Альдегиды могут быть окислены далее – до карбоновых кислот, в отличие
от кетонов, которые являются “тупиковой ветвью развития” и могут только снова стать вторичными спиртами.
Такой реакцией является взаимодействие многоатомного спирта со свежеприготовленным гидроксидом меди II. В результате реакции раствор
окрашивается в характерный синий цвет.
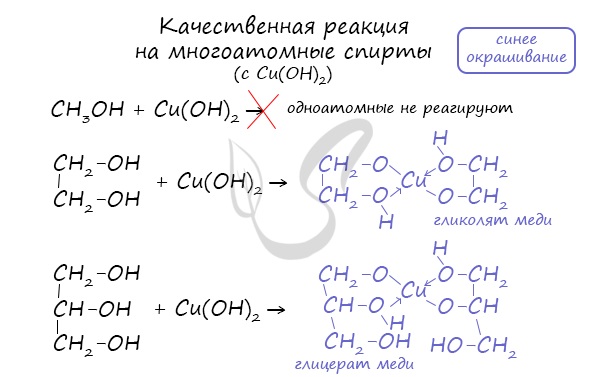
Важным отличием многоатомных спиртов от одноатомных является их способность реагировать со щелочами (что невозможно для одноатомных спиртов).
Это говорит об их более выраженных кислотных свойствах.
© Беллевич Юрий Сергеевич 2018-2023
Данная статья написана Беллевичем Юрием Сергеевичем и является его интеллектуальной собственностью. Копирование, распространение
(в том числе путем копирования на другие сайты и ресурсы в Интернете) или любое иное использование информации и объектов
без предварительного согласия правообладателя преследуется по закону. Для получения материалов статьи и разрешения их использования,
обратитесь, пожалуйста, к Беллевичу Юрию.
Спирты являются обширным и очень разнообразным классом органических соединений: они широко распространены в природе, имеют важнейшее промышленное значение и обладают исключительными химическими свойствами.
Существует огромное количество методов получения спиртов, при этом их можно разделить на две условных группы:
- химические способы получения спиртов — синтетические спирты;
- биохимические способы получения спиртов — биоспирты.
Химические способы получения спиртов[править | править код]
Занимая одну из центральных позиций в органической химии, спирты могут быть получены из множества других соединений.
На практике в качестве исходных веществ для синтеза спиртов наиболее часто используют [1][2] :
- алкилгалогениды — щелочной гидролиз или реакция с супероксидом калия;
- алкены — кислотная гидратация, реакция гидроксимеркурирования-демеркурирования или гидроборирование с последующим окислением, а также промышленные методы оксо-синтеза;
- карбонильные соединения — восстановление или взаимодействие с реактивами Гриньяра.
Далее в разделе будет подробно рассмотрена химия существующих методов получения одноатомных спиртов. Промышленные аспекты получения спиртов, включая биохимические методы синтеза, подробно рассмотрены в подразделе «Получение спиртов в промышленности».
Краткий обзор методов органического синтеза многоатомных спиртов будет рассмотрен в соответствующем подразделе.
Получение спиртов из галогеноуглеводородов[править | править код]
Галогенпроизводные углеводородов под действием оснований трансформируются с образованием спиртов (реакция нуклеофильного замещения).
Обычно, первичные и вторичные галогенуглеводороды вступают в реакцию по одностадийному SN2 механизму[3]. Пример — гидролиз бромэтана:

Реакции такого типа, обычно, происходят — с обращением геометрической конфигурации исходного вещества[3]. Реакционная способность алкинов уменьшается при переходе от производных йода к производным фтора [4] При этом фторпроизводные устойчивы к нуклеофильному замещению в обычных условиях и практически не используются для получения спиртов.
Первичные хлоралканы удовлетворительно гидролизуются под действием водного раствора щёлочи при нагревании[5]:

Для реакций, протекающих по SN2 механизму, используют только полярные растворители, причем скорость превращения возрастает при использовании вместо протонных растворителей (например: вода или спирт) апротонные (например: диметилсульфоксид); при этом в апротонных растворителях нуклеофильность уходящих групп будет иной[3]:

Третичные и в меньшей степени вторичные галогенуглеводороды гидролизуются по двухстадийному SN1 механизму[3]:
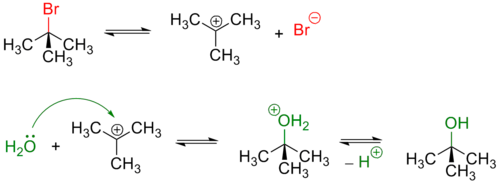
Реакция, протекающая по SN1 механизму проводят в полярных протонных растворителях, чаще всего воде или водном растворе метилового или этилового спирта.
Из-за устойчивости карбкатиона по такому механизму гидролизуются галогеналкены:

Так как в процессе реакции образуется карбкатион, его атака (в идеальных условиях без учёта фактора влияния заместителей) нуклеофилом может происходить с обеих сторон, что приводит к рацемизации образующегося продукта.
Для высокореакционных реагентов используют мягкое замещение с использованием соединений одновалентного серебра или двухвалентной ртути[5]:

![{displaystyle {mathsf {C_{6}H_{13}!!-!!CHBr!!-!!CH_{3}+H_{2}O}} {xrightarrow[{25^{o}C, CH_{3}OC_{2}H_{4}OCH_{3}}]{Hg(ClO_{4})_{2}}} {mathsf {C_{6}H_{13}!!-!!CH(OH)!!-!!CH_{3} _{(88%)}+HBr}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/643f43404fcb95e30c049dafabe0c13acf4b4477)
В современной лабораторной практике описанные выше реакции сольволиза проводят достаточно редко, так как спирты — более доступные полупродукты — являются исходным объектом для синтеза галогенпроизводных.
Кроме того, следует помнить, что помимо изменения стереохимии исходных компонентов, реакции замещения конкурируют с элиминированием, а также перегруппировками, что часто приводит к нежелательным продуктам[3]:


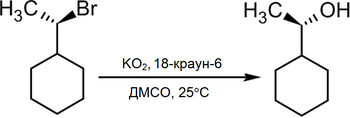
В то же время существует достаточно новый метод превращения в спирты алкилгалогенидов действием на последние супероксида калия в среде диметилсульфоксида в присутствии 18-краун-6, при этом происходит практически полное геометрическое обращение[2]:
Получение спиртов из алкенов[править | править код]
Гидратация алкенов[править | править код]
Кислотная гидратация алкенов исторически была первым синтетическим методом получения спиртов (см. подраздел «История открытия спиртов»).
Общий механизм процесса (реакция электрофильного присоединения AdE2) представлен ниже[6]:

![{mathsf {R!!-!!CH!!=!!CH_{2}+H_{3}O^{+}}}rightleftarrows {mathsf {[R!!-!!CH^{+}!!-!!CH_{3}]+H_{2}O}}rightarrow {mathsf {R!!-!!CH(OH)!!-!!CH_{3}+H^{+}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d328267dd281e15799995b90729af2a748ef526d)
Присоединение происходит по правилу Марковникова.
В случае использования серной кислоты в качестве катализатора промежуточным продуктом является эфир серной кислоты (R-CH(OSO2OH)-CH3), который в условиях реакции полностью гидролизуется до спирта[6].
Для проведения реакции кроме серной кислоты используют и другие реагенты: смесь муравьиной и каталитического количества серной кислоты (в отдельных случаях позволяет добиться стереоспецифичности), смесь муравьиной и хлорной кислоты, трифторуксуную кислоту и др[7].
Реакции вторичных алкенов, вследствие перегруппировок карбокатионов, часто приводят к образованию смеси продуктов, что затрудняет их использование для получения вторичных спиртов[8]:
![{mathsf {CH_{3}(CH_{2})_{3}CH!!=!!CH_{2}+H_{2}O}} {xrightarrow[ {85-100^{o}C}]{HCOOH, HClO_{4}}} {mathsf {CH_{3}(CH_{2})_{3}CH(OH)CH_{3}+CH_{3}(CH_{2})_{2}CH(OH)CH_{2}CH_{3}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/04382bada416065ffd5c1141a1f46a2388782eaf)
В лабораторной практике метод кислотной гидратации применим весьма ограниченно как из-за перспективы получения смеси продуктов, так и низких выходов. Чаще его используют для получения третичных спиртов, но и в этом случае выход, обычно, не превышает 40-45 %[8]:
![{displaystyle {mathsf {(}}{CH_{3})_{2}C!!=!!CH_{2}+H_{2}O} {xrightarrow[{10-20^{o}C}]{65%H_{2}SO_{4}}} {mathsf {(CH_{3})_{2}C(OH)CH_{3}}} _{(45%)}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b715302d49cbd0703414fb74e7113d1ee4ee394e)

В промышленности, помимо жидкофазной используют прямую газофазную гидратацию алкенов. В качестве катализаторов используется фосфорная кислота на твердом носителе при 200—300 °C и давлении 2-8 МПа; при этом выход спиртов достигает 95 %[9]:
![{mathsf {CH_{2}!!=!!CH_{2}+H_{2}O}} {xrightarrow[ {300^{o}C, 70-80 ATM}]{H_{3}PO_{4}/SiO_{2}}} {mathsf {CH_{3}CH_{2}OH}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b2c8c9eee5ff95e5e128eee08d10f3b756374b18)
![{mathsf {CH_{3}CH!!=!!CH_{2}+H_{2}O}} {xrightarrow[ {200^{o}C, 20-30 ATM}]{H_{3}PO_{4}/SiO_{2}}} {mathsf {CH_{3}CH(OH)CH_{3}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5e529dc175a1a28f3d2bf041bb254e7a4853b4b3)
Гидроксимеркурирование-демеркурирование алкенов[править | править код]
Метод получения спиртов гидроксимеркурированием-демеркурированием алкенов имеет ряд важных преимуществ перед реакцией кислотного гидролиза[10]:
- отсутствие перегруппировок для склонных для этого субстратов;
- анти-стереоспецифичность для нормальных алкенов за исключением особых случаев пространственного затруднения;
- лучшие выходы;
- строгая ориентация по правилу Марковникова.
Механизм реакции выглядит следующим образом[11]:

Присоединение ацетата ртути к алкену происходит по электрофильному механизму, а демеркурирование имеет радикальную природу; так как последняя стадия не обладает высокой стереоселективностью, то и весь процесс не стереоспецифичен в строгом смысле[12].
Синтез спиртов гидроксимеркурированием-демеркурированием алкенов протекает в мягких условиях, с выходами близкими к количественным (90-98 %) и практически без образования побочных продуктов; при этом промежуточное ртутьорганическое соединение не требует выделения — все стадии реакции протекают в один за другим[12].
Практические примеры использования реакции (в скобках указаны выходы, показывающие соотношение образующихся продуктов)[12]:
![{displaystyle {mathsf {CH_{3}(CH_{2})_{3}CH!!=!!CH_{2}}} {xrightarrow[{2) NaBH_{4}; H_{2}O}]{1) (CH_{3}COO)_{2}Hg,; THF-H_{2}O; 20^{o}C}} {mathsf {CH_{3}(CH_{2})_{3}CH(OH)CH_{3}}} _{(99,5%)}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7ca0165e755be7689f5a86d2758d414b1a2da3d0)
![{displaystyle {mathsf {C_{6}H_{5}!!-!!C(CH_{3})!!=!!CH_{2}}} {xrightarrow[{2) NaBH_{4}; H_{2}O}]{1) (CH_{3}COO)_{2}Hg,; THF-H_{2}O; 20^{o}C}} {mathsf {C_{6}H_{5}!!-!!C(CH_{3})(OH)!!-!!CH_{3}}} _{(100%)}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f296e2a5d0bfb1838eac9c83a38d72583280e9c8)
![{displaystyle {mathsf {(CH_{3})_{3}C!!-!!CH!!=!!CH_{2}}} {xrightarrow[{2) NaBH_{4}; H_{2}O}]{1) (CH_{3}COO)_{2}Hg,; THF-H_{2}O; 20^{o}C}} {mathsf {(CH_{3})_{3}C!!-!!CH(OH)!!-!!CH_{3}}} _{(97%)}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3435fa8d8414081f9f598cae2d36115b131012de)

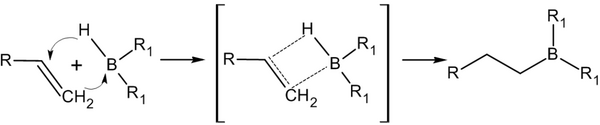
Гидроборирование алкенов с последующим окислением[править | править код]
Присоединение гидридов бора к алкенам и последующее их расщепление в щелочной среде, открытое Г. Брауном в 1958 году, является столь важной реакцией, что за её обнаружение и изучение в 1979 году учёный был удостоен Нобелевской премии по химии[13].
Присоединение происходит многоступенчато с образованием промежуточного циклического активированного комплекса, причем присоединение бора происходит против правила Марковникова — к наиболее гидрогенизированному атому углерода[14]:
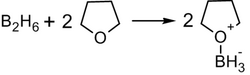
В синтезе используется, обычно, не собственно диборан, а его донорно-акцептоный комплекс с простым эфиром; а сам диборан получают реакцией борогидрида натрия с трифторидом бора в среде тетрагидрофурана[14]:

Алкилбораны легко расщепляются под действием пероксида водорода в щелочной среде, образуя спирты[14]:

Реакция гидроборирования является реакцией син-присоединения — её результатом становятся цис-аддукты.
Данный метод имеет широкое препаративное значение. Например, алкены с концевой двойной связью дают первичные спирты с выходами 80-90 %. Примеры практического использования метода (в скобках указаны выходы, показывающие соотношение образующихся продуктов)[7]:
![{displaystyle {mathsf {RCH!!=!!CH_{2}}} {xrightarrow[{H_{2}O_{2}, OH^{-}}]{B_{2}H_{6}}} {mathsf {RCH_{2}!!-!!CH_{2}OH}} _{(94%)}+{mathsf {RCH(OH)!!-!!CH_{3}}} _{(6%)}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/58c2b1070a75ab8dd5b19a5a192f9b06bf8d4ff1)
![{displaystyle {mathsf {C_{6}H_{5}CH!!=!!CH_{2}}} {xrightarrow[{H_{2}O_{2}, OH^{-}}]{B_{2}H_{6}}} {mathsf {C_{6}H_{5}CH_{2}!!-!!CH_{2}OH}} _{(80%)}+{mathsf {C_{6}H_{5}CH(OH)!!-!!CH_{3}}} _{(20%)}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/517b7f559eccc8bf0b46316c3b00fad22b0a5b66)
![{displaystyle {mathsf {(CH_{3})_{2}CHCH!!=!!CHCH_{3}}} {xrightarrow[{H_{2}O_{2}, OH^{-}}]{B_{2}H_{6}}} {mathsf {(CH_{3})_{2}CHCH_{2}CH(OH)CH_{3}}} _{(57%)}+{mathsf {(CH_{3})_{2}CHCH(OH)CH_{2}CH_{3}}} _{(43%)}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/81b80b2d3a569a3922217a4619e2735589d7d447)
Пример синтеза бициклических терпеновых спиртов[15]:

Для повышения селективности реакции гидроборирования используют замещённые, пространственно затруднённые бораны[16]:
Использование, например, в реакции со стиролом дисиамилборана (DIAB) повышает выход первичного спирта с 80 % до 98 %[17]:
![{displaystyle {mathsf {C_{6}H_{5}CH!!=!!CH_{2}}} {xrightarrow[{H_{2}O_{2}, OH^{-}}]{DIAB}} {mathsf {C_{6}H_{5}CH_{2}!!-!!CH_{2}OH}} _{(98%)}+{mathsf {C_{6}H_{5}CH(OH)!!-!!CH_{3}}} _{(2%)}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/71266e52684055ea9e54dacdfd61636e1d9f2196)
Высокая селективность указанных выше производных борана позволяет избирательно вступать в реакцию с цис-изомером, находящемся в смеси транс-изомером или гидроборировать одну двойную связь из двух имеющихся в молекуле алкена, например[6]:
![{mathsf {CH_{2}!!=!!CH!!-!!(CH_{2})_{3}!!-!!C(CH_{3})!!=!!CH_{2}}} {xrightarrow[ {H_{2}O_{2}, OH^{-}}]{(Sia)_{2}BH}} {mathsf {HOCH_{2}!!-!!CH_{2}!!-!!(CH_{2})_{3}!!-!!C(CH_{3})!!=!!CH_{2}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/512dad8f188aab6338939c59597445cfb7f40dd1)
Гидроборирование алкенов с последующим присоединением окиси углерода[править | править код]
Одним из лучших способов получения третичных спиртов является присоединение к окиси углерода к алкилборанам. Реакция легко протекает при обычном давлении и температуре около 125 °C, в качестве растворителя используется диглим[7]:

![{mathsf {(RCH_{2}!!-!!CH_{2}-)_{3}B+CO}}{xrightarrow {125^{o}C}}{mathsf {[(RCH_{2}!!-!!CH_{2}-)_{3}B^{-}!!!!-!!C}}!!equiv !!{mathsf {O^{+}]}}rightarrow {mathsf {[(RCH_{2}!!-!!CH_{2}-)_{2}B!!-!!CO!!-!!CH_{2}CH_{2}R]}}rightarrow](https://wikimedia.org/api/rest_v1/media/math/render/svg/5c321c4c32849482f1301a2b54d802a31e86a612)
![rightarrow {mathsf {[(RCH_{2}!!-!!CH_{2}-)_{3}C!!-!!B!!=!!O]}}{xrightarrow[ {NaOH, H_{2}O}]{H_{2}O_{2}}}{mathsf {(RCH_{2}!!-!!CH_{2}-)_{3}COH+Na[B(OH)_{4}]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/211637566b6dbbd36c8966d5f1231d963b04545b)
Этот метод даёт неплохие выходы со многими алкенами[14]:
![{displaystyle {mathsf {CH_{3}CH!!=!!CHCH_{3}}}{xrightarrow[{3) H_{2}O_{2}, NaOH, H_{2}O}]{1) B_{2}H_{6}; 2) CO}}{mathsf {(CH_{3}CH_{2}CH(CH_{3})-)_{3}COH}} _{(87%)}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/acfb6d0a6d5c002959880578fa3022eb9dda40ce)

Если данную реакцию проводить в присутствии водного раствора щёлочи, получаются вторичные спирты[18]:

Гидроформилирование алкенов[править | править код]
Широко используемая в промышленности классическая реакция гидроформилирования алкенов, то есть каталитического присоединение к ним водорода и монооксида углерода с получением на выходе альдегидов[19], может быть проведена таким образом, что продуктом её реакции сразу будут спирты без выделения промежуточных карбонильных соединений Это метод иногда называют восстановительным гидроформилированием.

Катализирует реакцию координационно ненасыщенный гидрокарбонил кобальта, образующийся в ходе реакции[19]:


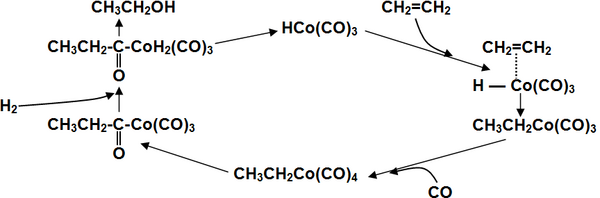
Для получения спиртов в одну стадию в качестве катализаторов используют карбонилы кобальта, модифицированные фосфинами, что помимо более активного гидрирования, позволяет добиться существенно более высокой селективности выхода нормальных продуктов (до 90 %) вследствие стерического эффекта объёмного фосфинового лиганда в переходном состоянии[20].
Получение спиртов из простых эфиров и спиртов[править | править код]
Реакция гомологизации спиртов[править | править код]
Гомологизация, то есть превращение органического соединения в свой гомолог путём внедрения одной или нескольких метиленовых групп, для спиртов была впервые осуществлена в 1940 году — на основе метанола каталитическим путём под воздействием высокого давления был синтезирован этанол[21]:

Реакция гомологизации по своему механизму близка реакции гидроформилирования алкенов и в настоящее время с помощью модифицированных катализаторов кобальта и рутения и добавления йодид-ионов в качестве промоторов удаётся добиться 90 % выхода по этанолу[21].
Исходный метанол также получают из окиси углерода (катализаторы на основе оксидов меди и цинка, давление 5-10 МПа, температура 250 °C)[21], так что общая схема выглядит следующим образом:
![{mathsf {C+H_{2}O}}rightarrow {mathsf {CO+H_{2}}}rightarrow {mathsf {CH_{3}OH}} {xrightarrow[ {- H_{2}O}]{CO + H_{2}}} {mathsf {CH_{3}CH_{2}OH}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f2e3410591ebad2e5821584fa4fc68aad8d10200)
Побочными продуктами реакции в случае синтеза этанола будут ацетальдегид, этилен и диэтиловый эфир.
Реакция Гербе́[править | править код]
Реакция Гербе представляет собой высокотемпературный (200 °C, давление 5—6 МПа) процесс каталитической конденсации первичных алифатических спиртов, не имеющих разветвления в α-положении, по следующей схеме[22]:

В качестве катализаторов используют сложную смесь на основе никеля Ренея, меди, солей железа и других компонентов[23].
Предполагаемый условный механизм реакции[23]:



или

Реакция имеет ограниченное применение как из-за жестких условий её проведения и относительно низкого выхода (как правило, до 70 %), так и образования кислоты и альдегида в качестве побочного продукта[23].
Кислотное расщепление простых эфиров[править | править код]
В лабораторной практике подобный способ получения спиртов крайне редок, так как именно спирты служат исходным компонентом для синтеза простых эфиров. Вместе с тем, если в качестве исходного объекта выбран, скажем природный простой эфир сложной структуры, его лабораторное расщепление до исходного спирта может оказаться востребованным. Кроме того, в некоторых случаях для защиты гидроксильной группы в процессе многоступенчатого синтеза, её могут перевести в эфирную и вводить в реакцию уже простой эфир. По окончании процесса для обратного превращения соединения в спирт может потребоваться расщепление эфира (см. подробнее подраздел «Защита через простые эфиры»).
Обычно реакцию проводят нагреванием эфира и концентрированного раствора бромоводородной или йодоводородной кислоты, при этом расщепление может осуществляться как по механизму SN1, так и SN2[24]:
|
SN1 механизм расщепления простых эфиров |
SN2 механизм расщепления простых эфиров |


Если в реакцию вступают несимметричные эфиры, в результате получаются два спирта и два галогенпроизводных, однако если эфир метиловый, продуктом реакции будет спирт и метилиодид или метилбромид[25]:

Для расщепления эфиров могут использоваться также кислоты Льюиса: BF3, BCl3, AlCl3 и др[25], а также сильные органические кислоты. Например, расщепление трет-бутилциклогексилового эфира трифторуксуной кислотой происходит по механизму SN1 с образованием циклогексанола и 2-метилпропена[26]:

Перегруппировка Виттига[править | править код]
Простые эфиры под действием фениллития перегруппировываются в спирты (Георг Виттиг, 1942 год):

Реакция представляет собой карбанионную перегруппировку, которая осуществляется через радикальный механизм расщепления-рекомбинации[27]:
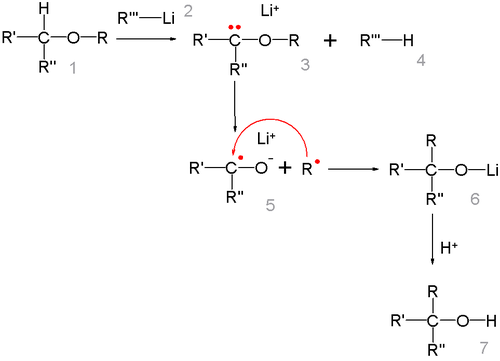
Говоря о стереохимии перегруппировки Виттига, следует отметить, что образование новой C-C связи происходит настолько быстро, что радикал R не успевает инвертироваться, поэтому обычно, реакция протекает с сохранением исходной конфигурации[28].
Изучение перегруппировки на примере β-алкоксиалкилаллиловых эфиров (общий вид:  ) показало, что в результате реакции с выходом 14-32 % образовывались син-1,3-диол производные с селективностью 90-95 %[29].
) показало, что в результате реакции с выходом 14-32 % образовывались син-1,3-диол производные с селективностью 90-95 %[29].
Перегруппировка Виттига может осуществляться не только с помощью алкил- или ариллитиевых соединений (фениллитий, бутиллитий, метиллитий, диэтиламид лития и пр.), но и под действием других сильных оснований; например, следующая реакция протекает в жидком аммиаке в присутствии амида калия (выход 90 %)[30]:

Для аллильно-замещённых субстратов (1,2)-перегруппировка конкурирует с (2,3)-перегруппировкой, которую можно наблюдать практически полностью независимо при низких температурах[27]:

Получение спиртов из альдегидов и кетонов[править | править код]
В данном разделе помимо получения собственно спиртов из альдегидов и кетонов приведены синтезы гидроксикарбонильных соединений (кетоспирты и производные гидроксикарбоновых кислот; смотри подразделы «Альдольная конденсация», «Бензоиновая конденсация», «Реакция Реформатского», «Реакция Иванова»). Это связано с тем, что приведённые реакции являются мощными препаративными методами и широко используются на практике.
Этинилирование карбонильных соединений[править | править код]
Важным методом получения ацетиленовых спиртов является реакция Фаворского, иначе говоря, реакция этинилирования карбонильных соединений:


В реакцию вступают незамещённые алкины (берутся в большом избытке), кетоны и некоторые альдегиды (чаще всего используется формальдегид) в присутствии оснований (KOH или NaNH2 в органическом растворителе) при температуре от −70 до +40 °C, давлении 0,4-0,9 МПа[31].
Механизм данной реакции связан с нуклеофильным присоединеним этинильного карбаниона к карбонильной группе[32]:
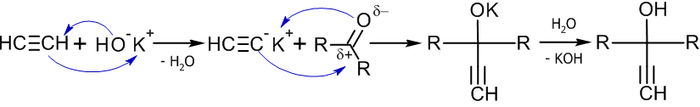
Примеры реакций[33]:


Существуют, по-меньшей мере, две модификации этого метода:
- Реакция Реппе — конденсация алкинов с альдегидами или кетонами в присутствии каталитических количеств ацетиленидов меди, серебра или ртути[34].
- Реакция Нефа — реакция с последующим гидролизом ацетиленидов щелочных металлов с кетонами, включая α,β-непредельные и ароматические карбонильные соединения[35].
Взаимодействие альдегидов с аллилборанами[править | править код]
Современным методом получения аллильных спиртов заданной конфигурации является применение в качестве агента аллилборана, который реагирует с альдегидами в присутствии оснований по следующей схеме[36]:

Реакция была использована, в частности, в качестве базовой для полного синтеза ряда природных
соединений и их аналогов, например, феромонов короеда[36]:
Существуют различные методики этой реакции, среди которых:
- Присоединение кротилборана к альдегидам (Хоффман, 1982 год)[37]:
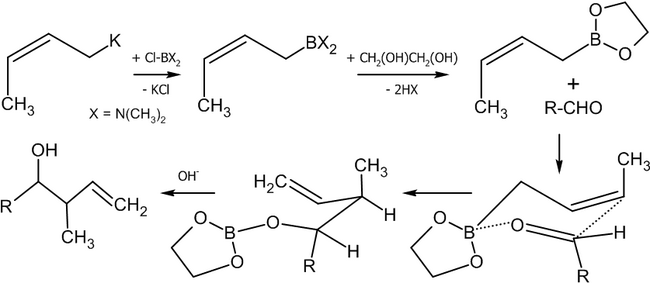
Если в реакцию вступают цис-алкены, в основном, будут образовываться син-продукты присоединения (97 % от общего выхода).
Другим удобным агентом, реагирующим по данной схеме является аллилборный эфир винной кислоты (Рауш, 1985 год)[38].
- Региоселективный синтез линейных или разветвлённых аллиловых спиртов (Ямамото, 1983 год)[37]:

Как видно из схемы, меняя температуру реакции, можно вызвать миграцию атома бора к соседнему углероду, получив тот или иной изомерный спирт.
Реакция Сакураи[править | править код]
Другой способ аллилирования, заключающийся в электрофильном взаимодействии аллилсиланов с различными соединениями в присутствии кислот Льюиса носит название реакции Сакураи. С точки зрения получения спиртов, существуют две модификации подобного синтеза[39]:
- Реакция с карбонильными соединениями с получением вторичных или третичных спиртов:
![{mathsf {RCOR'+CH_{2}!!=!!CHCH_{2}Si(CH_{3})_{3}}} {xrightarrow[ {H_{2}O}]{Lewis acid}} {mathsf {RR'C(OH)CH_{2}CH!!=!!CH_{2}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a6c420605cd1d7b79587a1becd220bfd02155530)
- Реакция с эпоксидами с получением вторичных спиртов:
![{mathsf {(CHR!!-!!CHR)O+CH_{2}!!=!!CHCH_{2}Si(CH_{3})_{3}}} {xrightarrow[ {H_{2}O}]{Lewis acid}} {mathsf {RCH(OH)CH(R)CH_{2}CH!!=!!CH_{2}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c7850c9b031fda24164c2f838fc3dc26ff98e352)
В качестве катализаторов реакции могут выступать: TiCl4, BF3, SnCl4 и пр.
Пример практического использования реакции Сакураи[40]:
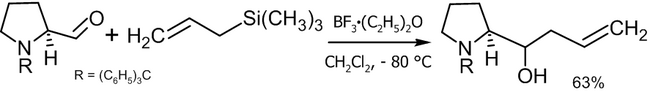
Реакция Бэйлиса-Хиллмана-Морита[править | править код]
Традиционная реакция Бэйлиса-Хиллмана-Морита представляет собой метод получения аллиловых кетоспиртов взаимодействием альдегидов с метилвинилкетонами или другими активированными алкенами в присутствии третичных фосфинов и каталитических количеств фенола или его производных[41]. Впоследствии реакция была несколько модифицирована: в качестве катализатора стали использовать третичные амины (например: 1,4-диазобицикло[2.2.2]октан или DABCO[42]):

Предположительно, механизм реакции выглядит следующим образом[43]:

Реакция Нозаки-Хияма-Киши[править | править код]
Реакция Нозаки-Хияма-Киши представляет собой современный метод получения спиртов селективным восстановительным сочетанием альдегидов с винил- или аллилгалогенидами (бромиды или йодиды) в присутствии хром-никелевого катализатора[44]:
![{mathsf {R'!!-!!CHO+R!!-!!CH!!=!!CHBr}} {xrightarrow[ {(CH_{3})_{2}S=O}]{CrCl_{2}, NiBr_{2}}} {mathsf {R!!-!!CH!!=!!CH!!-!!CH(OH)R'}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4a98eec60089d57d2df1fa9c60c94de1d47606c2)
Каталитический цикл реакции выглядит следующим образом[45]:

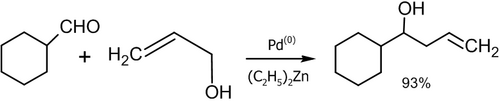
Каталитическое сочетание альдегидов с аллиловыми спиртами и их производными[править | править код]
Аналогом синтетического метода, рассмотренного в предыдущем подразделе, является реакция сочетания альдегидов с аллиловыми спиртами и их производными в присутствии катализаторов. В научной литературе описано множество лабораторных методик осуществления подробного синтеза с примененением органических соединений кремния, олова, хрома, лития, рутения, палладия, цинка, титана, циркония и других металлов.
Приведём некоторые характерные примеры использования этого метода на практике:
- Реакция аллилацетата с альдегидами в присутствии солей рутения (Denmark S. E., Nguyen S. T., 2009 год)[46]:

- Реакция аллилового спирта с алифатическими альдегидами в присутствии палладиевого катализатора (Masanari Kimura, Masamichi Shimizu, Kazufumi Shibata, Minoru Tazoe, Yoshinao Tamaru, 2003 год)[47]:
- Реакция аллилтрибутилстанната с альдегидами в присутствии рениевого комплекса (Yutaka Nishiyama, Fujio Kakushoua, Noboru Sonoda, 2004 год)[48]:
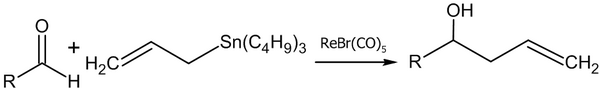
Более подробно о современных методах получения спиртов реакцией аллиловых спиртов и их производных с карбонильными соединениями можно прочитать в монографии: Junzo Otera. Modern Carbonyl Chemistry — Wiley-VCH, Weinheim, 2000—613 Pages — ISBN 978-3-527-29871-6.
Реакция Канниццаро[править | править код]
Реакция Канниццаро представляет собой оксилительно-восстановительное диспропорционирование альдегидов в первичные спирты и карбоновые кислоты под действием оснований[49]:

На первом этапе реакции происходит нуклеофильное присоединение основания (например: гидроксид-аниона) к карбонильному углероду альдегида. Образующийся анион депротонируется (это требует воздействия достаточно сильного основания) с образованием промежуточного дианиона, который затем вступает в реакцию с молекулой альдегида:

В реакцию Канниццаро вступают альдегиды, не способные к енолизации (не имеющие α-водорода), поскольку для последних преобладающей будет альдольная конденсация. Например, в известном примере с бензальдегидом, выход бензилового спирта может достигать 90 %[50]:

Чаще реакция Канниццаро используется для синтеза ароматических и гетероароматических спиртов[50].
Для повышения реакционного диапазона используемых альдегидов и увеличения выхода спиртов на практике используется перекрестная реакция Канниццаро, то есть использование двух различных альдегидов, причем в качестве альдегида-восстановителя обычно используется формальдегид, который в ходе реакции окисляется в муравьиную кислоту[51]:

В настоящее время существуют более эффективные синтетические методы, поэтому полезность реакции Канниццаро ограничена, как правило, диспропорционированием кетоальдегидов в гидроксикарбоновые кислоты[52]:

Циангидринный синтез[править | править код]
Карбонильные соединения, особенно альдегиды и стерически не затруднённые кетоны, легко вступают в реакции нуклеофильного присоединения c цианистым водородом (нуклеофил CN−) с образованием циангидринов[53]:

Для ароматических кетонов вместо HCN используют цианид диэтилалюминия (C2H5)2AlCN или цианотриметилсилан (CH3)3SiCN, продукт присоединения которого затем гидролизуется до циангидрина[53]:

Далее при необходимости циангидрин легко гидролизуется до гидроксикислоты или восстанавливается в аминоспирт:


Альдольная конденсация[править | править код]
Альдольная конденсация — это одна из старейших реакций органического синтеза (1872 год, Вюрц), в которой две молекулы альдегида или кетона под действием основания или кислоты соединяясь, образуют кетоспирты или альдоли[54]:

Возможны два механизма этой реакции: щелочной или кислотный, однако с точки зрения синтеза спиртов, последний менее предпочтителен, так как часто реакция не останавливается на стадии спирта а протекает дальше с дегидратацией и образованием непредельных карбонильных соединений (кротоновая конденсация)[54].
Механизм конденсации, происходящий под действием основания, следующий[54]:

Для проведения конденсации, как видно из её механизма, необходимо, чтобы хотя бы одна из молекул содержала водород в α-положении к карбонильной группе. Обычно, для отрыва этого атома водорода силы гидроксид-иона бывает достаточно, но в отдельных случаях используют и более сильные основания, например — бутиллитий.
Возможны пять комбинаций протекания альдольной конденсации[53]:
- Взаимодействие двух молекул одного альдегида: реакция легко осуществима и приводит к одному продукту, однако из-за электроноакцепторного эффекта альдегидной группы, использование их для синтеза спиртов не эффективно из-за обычно преобладающего расщепления образующегося альдоля до непредельного альдегида, например:

- Взаимодействие двух молекул различных альдегидов: теоретически реакция может привести к четырём разным продуктам, но если один из альдегидов не будет содержать α-карбонильный водород, возможно осуществление только перекрестной реакции;
- Взаимодействие двух молекул одного кетона: реакция сильно смещена влево, поэтому для её проведения либо пользуются специальным оборудованием (аппарат Сокслета), позволяющим фактически удалять из реакционной зоны продукт реакции или использовать в качестве основания особые реагенты (например: нитрид бария);
- Взаимодействие двух молекул разных кетонов: применяется довольно редко и в случаях, когда один из кетонов не содержит α-карбонильный водород;
- Взаимодействие одной молекулы альдегида с одной молекулой кетона: чаще всего в качестве альдегида используют формальдегид, который даёт один продукт конденсации. Другой вариант — использование не самого кетона, а его енольной формы, например в виде литиевой соли или силилового эфира.
Кроме собственно альдегида, возможно использование имина и диизопропиламида лития в качестве основания[53]:

Разновидностью альдольной реакции является реакция Мукаямы, в которой используются силиленольные эфиры:

Бензоиновая конденсация[править | править код]
Бензоиновая конденсация представляет собой обратимое образование из альдегидов (преимущественно — ароматических) под действием цианид-ионов CN− α-оксикетонов (ацилоинов) с общей формулой —СR(ОН)—С(O)О—:

В этой реакции на первом этапе цианид анион вступает в реакцию нуклеофильного присоединения с альдегидом. Перегруппировка образующегося интермедиата в карбанион завершается дальнейшим присоединением второй карбонильной группы также по нуклеофильному механизму. Завершает реакцию переноса протона и отщепление цианидной группы с образованием бензоина в качестве конечного продукта.
Реакция Иванова[править | править код]
Реакция Иванова представляет собой метод получения β-гидроксикарбоновых кислот общей формулой —CR(OH)—CR1(COOH)— из карбонильных соединений и реактивов Иванова: магнийгалогенпроизводных солей арилуксусных кислот (обычно — фенилуксусной кислоты)[55]. Например:
![{mathsf {R!!-!!CO!!-!!R+[Ar!!-!!CH^{-}!!!!-!!COONa]MgCl^{+}}}rightarrow {mathsf {[Ar!!-!!CR(COONa)!!-!!CHR(OMgCl)]}}rightarrow {mathsf {Ar!!-!!CR(COOH)!!-!!CHROH}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ac338f015e3b2ba9bb5f4c364b3fcdeb6a9fe005)
При необходимости оксикислота может быть легко превращена в соответствующий спирт методом декарбоксилирования.
Получение спиртов из карбоновых кислот и сложных эфиров[править | править код]
Гидролиз сложных эфиров карбоновых кислот[править | править код]
Гидролиз сложных эфиров карбоновых кислот является типичной реакцией алифатического нуклеофильного замещения, проходящей по следующей условной схеме:

Обычно реакцию проводят при нагревании в щелочной среде. В частности, данный способ является одним из промышленных путей получения глицерина из животных или растительных жиров.
Ацилоиновая конденсация[править | править код]
Ацилоиновая конденсация представляет собой получение α-гидроксикетонов (ацилоинов) восстановлением сложных эфиров металлическим натрием:
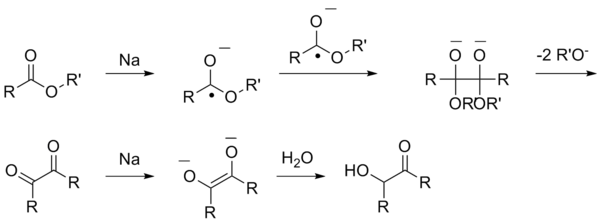
Данная реакция включает в себя несколько стадий, при которых сначала образуются анион-радикалы, а после ряда трансформаций алкоголяты, которые под действием воды переходят в ацилоины.
Декарбонилирование карбоновых кислот[править | править код]
Декарбонилирование (отщепление CO) карбоновых кислот достаточно редкий лабораторный способ получения спиртов, который может быть осуществлён с использованием металлических катализаторов.
Получение спиртов восстановлением эпоксидов и карбонильных соединений[править | править код]
Восстановление эпоксидов гидридами металлов[править | править код]
Восстановление карбонильных соединений гидридами металлов[править | править код]
Восстановление карбонильных соединений по реакции Меервейна-Пондорфа-Верлея[править | править код]
Восстановление карбонильных соединений органическими реагентами[править | править код]
Восстановление ароматических кетонов щелочными металлами[править | править код]
Восстановление карбоновых кислот и сложных эфиров по методу Буво-Блана[править | править код]

Восстановление хлорангидридов карбоновых кислот[править | править код]
Каталитическое гидрирование карбонильных соединений[править | править код]
Получение спиртов с использованием металлорганических соединений[править | править код]
Присоединение реактивов Гриньяра к эпоксидам[править | править код]
Присоединение реактивов Гриньяра к альдегидам или кетонам[править | править код]

Присоединение реактивов Гриньяра к сложным эфирам или ацилгалогенидам[править | править код]
Получение спиртов окислительными методами[править | править код]
Окисление алканов и циклоалканов[править | править код]
Окисление алкенов[править | править код]
Многоатомные спирты можно получить путём мягкого окисления (по Вагнеру) алкенов – для этого необходимо пропустить их через водный раствор окислителя, например – перманганата калия, – при температуре от 0 до 5 градусов Цельсия:

Также возможно и получение вторичных спиртов методом жёсткого окисления разветвлённых алкенов (либо подкисленным раствором окислителя, либо оксидом осмия (VIII)) с последующим восстановлением:

Озонолиз алкенов с последующим восстановлением[править | править код]
Спирт можно получить озонированием алкена с дальнейшей реакцией с сильным восстановителем (тетрагидроборат натрия, тетрадигидроалюминат лития):

Реакцию озонолиза следует проводить с использованием таких органических растворителей, как хлористый метилен или этилацетат[56].
Реакция Циглера[править | править код]
Является методом синтеза высших спиртов (С8 и выше) при помощи алюминийорганических соединений. Алюминийорганические соединения легко могут быть получены из олефинов в присутствии водорода. Данным методом можно получать чистые первичные спирты:
![{mathsf {RCH!!=!!CH_{2}}} {xrightarrow[ {-RH}]{H_{2}, AlR_{3}}} {mathsf {RCH_{2}CH_{2}!!-!!AlR_{2}}} {xrightarrow {O_{2}}} {mathsf {RCH_{2}CH_{2}!!-!!O!!-!!Al(OR)_{2}}} {xrightarrow {H_{2}O}} {mathsf {RCH_{2}CH_{2}OH}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/754c67e54bba7c18c2fb0a982958ff4d2f2bb257)
Окисление реактивов Гриньяра[править | править код]

Окисление Тамао-Кумада-Флеминга[править | править код]
Прочие методы получения спиртов[править | править код]
Гидролиз эфиров неорганических кислот[править | править код]
Диазотирование первичных алифатических аминов[править | править код]
Реакция Демьянова[править | править код]
Реакция Фриделя-Крафтса[править | править код]

Восстановление сложных эфиров тиокислот[править | править код]
Реакция Кулинковича[править | править код]
Краткий обзор методов органического синтеза многоатомных спиртов[править | править код]
Получение 1,2-диолов гидратацией эпоксидов[править | править код]
Получение 1,2-диолов конденсацией карбонильных соединений[править | править код]
Получение 1,2-диолов цис-дигидроксилированием алкенов[править | править код]
Получение 1,2-диолов транс-дигидроксилированием алкенов[править | править код]
Получение 1,3-диолов по реакции Принса[править | править код]
Получение многоатомных спиртов по реакции Толленса[править | править код]
Получение спиртов в промышленности[править | править код]
Получение спиртов методами основного органического синтеза[править | править код]
Промышленные синтезы на основе окиси углерода[править | править код]
Промышленное получение спиртов гидратацией алкенов[править | править код]
Промышленное получение спиртов окислением углеводородов[править | править код]
Промышленное получение спиртов гидрированием карбонильных соединений[править | править код]
Промышленное получение спиртов щелочным гидролизом галогенуглеводородов[править | править код]
Химические и биохимические методы получения спиртов из природного сырья[править | править код]
Промышленное получение спиртов щелочным гидролизом природного сырья[править | править код]
Биохимическое гидролизное получение спиртов[править | править код]
Промышленный биосинтез спиртов[править | править код]
Примечания[править | править код]
- ↑
Спирты // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 4. — С. 800. - ↑ 1 2 Курц А.Л., Брусова Г.П., Демьянович В.М. Получение одноатомных спиртов. Одно- и двухатомные спирты, простые эфиры и их сернистые аналоги. ChemNet. Химический факультет МГУ (1999). Дата обращения: 1 сентября 2009. Архивировано 29 апреля 2012 года.
- ↑ 1 2 3 4 5
Травень В.Ф. Органическая химия: Учебник для вузов: В 2 т. / В.Ф.Травень. — М.: ИКЦ «Академкнига», 2004. — Т. 1. — 586-623 с. — ISBN 5-94628-171-2. - ↑ Глава 3.3.3. Нуклеофильное замещение галогена // Общая органическая химия. Стереохимия, углеводороды, галогенсодержащие соединения = Comprehensive Organic Chemistry / Под ред. Д.Бартона и В.Д.Оллиса. — М.: «Химия», 1981. — Т. 1. — С. 660-661.
- ↑ 1 2 Глава 4.1.1. Одноатомные спирты // Общая органическая химия. Кислородсодержащие соединения = Comprehensive Organic Chemistry / Под ред. Д.Бартона и В.Д.Оллиса. — М.: «Химия», 1982. — Т. 2. — С. 13-118.
- ↑ 1 2 3
Mарч Дж. Глава 15. Реакции присоединения к кратным связям углерод-углерод // Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: «Мир», 1988. — Т. 3. — С. 132-212. - ↑ 1 2 3
Бюлер К., Пирсон Д. Глава 4. Спирты. Б. Реакции присоединения и замещения // Органические синтезы = Survey of Organic Syntheses / Пер. с англ. — М.: «Мир», 1973. — Т. 1. — С. 213-219. - ↑ 1 2 Курц А Л., Ливанцов М.В., Ливанцова Л.И. Гидратация алкенов. Алкены (Часть 1). ChemNet. Химический факультет МГУ (1998). Дата обращения: 2 сентября 2009. Архивировано 4 марта 2016 года.
- ↑
Лебедев. Химия и технология основного органического и нефтехимического синтеза: Учебник для вузов / Пер. с англ. — 4-е изд. перераб. и доп.. — М.: «Химия», 1988. — С. 180-184. — ISBN 5-7245-0008-6. - ↑ Глава 2.2.3. Реакции олефинов // Общая органическая химия. Стереохимия, углеводороды, галогенсодержащие соединения = Comprehensive Organic Chemistry / Под ред. Д.Бартона и В.Д.Оллиса. — М.: «Химия», 1981. — Т. 1. — С. 207-208.
- ↑ Carey F.A. Oxymercuration-Demercuration of Alkenes (англ.). Organic chemistry. McGraw-Hill Higher Education. Дата обращения: 2 сентября 2009. Архивировано 23 апреля 2012 года.
- ↑ 1 2 3 Реутов О.А, Курц А.Л., Бутин К.П. Органическая химия. — М.: Издательство МГУ, 1999. — Т. 1. — С. 385-387. — ISBN 5-211-03054-0.
- ↑ The Nobel Prize in Chemistry 1979 (англ.). Nobel Prize in Chemistry. The Official Web Site of the Nobel Foundation. Дата обращения: 3 сентября 2009. Архивировано 22 августа 2011 года.
- ↑ 1 2 3 4 Курц А Л., Ливанцов М.В., Ливанцова Л.И. Гидроборирование алкенов. Алкены (Часть 2). ChemNet. Химический факультет МГУ (1998). Дата обращения: 2 сентября 2009. Архивировано из оригинала 19 декабря 2011 года.
- ↑ Кучин А.В., Фролова Л.Л., Пантелеева М.В. Бициклические терпеновые диолы как лиганды для синтеза хиральных катализаторов (pdf) (недоступная ссылка — история). Англо-русскоязычный общественный химический журнал
«Бутлеровские сообщения». Дата обращения: 31 августа 2009. Архивировано 23 апреля 2012 года. - ↑
Борорганические соединения // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 594-603. - ↑
Вацуро К.В., Мищенко Г.Л. 109. Браун (Brown H.C.) // Именные реакции в органичнеской химии. — М.: «Химия», 1976. — С. 76-77. - ↑
Дядченко В.П., Трушков И.В., Брусова Г.П. Синтетические методы органической химии. Части 1-2. — М.: МГУ им. М.В. Ломоносова, 2004. — С. 46. - ↑ 1 2 Караханов Э.А. Синтез-газ как альтернатива нефти. I. Процесс Фишера-Тропша и оксо-синтез // Соросовский образовательный журнал. — 1997. — № 3. — С. 73-74. (недоступная ссылка)
- ↑ Шелдон Р.А. Химические продукты на основе синтез-газа = Chemicals From Synthesis Gas / Под ред. С.М.Локтева. — М.: «Химия», 1987. — С. 92.
- ↑ 1 2 3 Караханов Э.А. Синтез-газ как альтернатива нефти. II. Метанол и синтезы на его основе // Соросовский образовательный журнал. — 1997. — № 12. — С. 68. (недоступная ссылка)
- ↑
Гербе реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 1024-1025. - ↑ 1 2 3
Бюлер К., Пирсон Д. Глава 4. Спирты. Ж. Присоединение карбанионов // Органические синтезы = Survey of Organic Syntheses / Пер. с англ. — М.: «Мир», 1973. — Т. 1. — С. 268-279. - ↑
Травень В.Ф. Глава 18. Простые эфиры. Циклические эфиры // Органическая химия: Учебник для вузов: В 2 т. / В.Ф.Травень. — М.: ИКЦ «Академкнига», 2004. — Т. 2. — 97-98 с. — ISBN 5-94628-172-0. - ↑ 1 2
Mарч Дж. Глава 10. Реакции алифатического нуклеофильного замещения // Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: «Мир», 1988. — Т. 2. — С. 11-240. - ↑ McMurry J. Organic chemistry. — Seven edition. — Thomson, 2008. — P. 604, 658. — ISBN 0-495-11258-5.
- ↑ 1 2 (1,2)-Wittig Rearrangement (англ.). Name Reactions. Organic Chemistry Portal. Дата обращения: 14 сентября 2009. Архивировано 23 апреля 2012 года.
- ↑ Бутин К.П. Механизмы органических реакций: достижения и перспективы // Журнал Российского химического общества им. Д.И.Менделеева. — 2001. — Т. XLV, № 2. — С. 32.
- ↑ Schreiber S.L., Goulet M. T. Stereochemistry of the 1,2-Wittig Rearangement: A Synthesis of syn-1,3-diol Monoethers (англ.) // Tetrahedron Letters. — 1987. — Vol. 28, no. 10. — P. 1043-1046.
- ↑ Физер Л., Физер М. Реагенты для органического синтеза = Reagents For Organic Synthesis / Под редакцией проф. И.Л.Кнунянца и д.х.н. Р.Г.Костяновского. — М.: «Мир», 1970. — Т. 1. — С. 56-57.
- ↑
Фаворского реакции // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 5. — С. 95-96. - ↑ Темкин О. Н. Химия ацетилена. «Ацетиленовое дерево» в органической химии XXI века // Соросовский образовательный журнал. — 2001. — Т. 7, № 6. — С. 39. (недоступная ссылка)
- ↑
Вацуро К.В., Мищенко Г.Л. 595. Фаворский // Именные реакции в органической химии. — М.: «Химия», 1976. — С. 411-412. - ↑
Вацуро К.В., Мищенко Г.Л. 506. Реппе (Reppe) // Именные реакции в органической химии. — М.: «Химия», 1976. — С. 351. - ↑
Вацуро К.В., Мищенко Г.Л. 424. Неф (Nef) // Именные реакции в органической химии. — М.: «Химия», 1976. — С. 296. - ↑ 1 2 Бубнов Ю.Н. Аллилбораны. Принципы реагирования и применение в органическом синтезе // Вестник Московского университета : Серия 2. Химия. — 2005. — Т. 46, № 3. — С. 140-144.
- ↑ 1 2 Reich I.L. Allylborane Reactions (англ.). Chemistry 842 – Fall 2004 Course Outline. University of Wisconsin. Department of Chemistry. Дата обращения: 15 сентября 2009.
- ↑ Ли Дж. Роуш (Roush). Аллилборонат как реагент // Именные реакции. Механизмы органических реакций = Name reactions / Пер. с англ. В.М.Демьянович. — М.: БИНОМ. Лаборатория знаний, 2006. — С. 306. — ISBN 5-94774-368-X.
- ↑ Hosomi-Sakurai Reaction (англ.). Name Reactions. Organic Chemistry Portal. Дата обращения: 20 сентября 2009. Архивировано 23 апреля 2012 года.
- ↑ Li J. J., Limberakis C., Pflum D. A. Modern organic synthesis in the laboratory: a collection of standard
experimental procedures. — New York: Oxford University Press, Inc, 2007. — P. 139. — ISBN 978-0-19-518798-4. - ↑ Traditional Morita-Baylis-Hillman reaction of aldehydes with methyl vinyl ketone co-catalyzed by triphenylphosphine and nitrophenol (англ.). Abstracts. Organic Chemistry Portal. Дата обращения: 23 сентября 2009. Архивировано 23 апреля 2012 года.
- ↑ Baylis-Hillman Reaction (англ.) (недоступная ссылка — история). Name Reactions. Organic Chemistry Portal. Дата обращения: 23 сентября 2009. Архивировано 21 августа 2011 года.
- ↑ Smith A.C. Morita Baylis Hillman Reaction (англ.) (pdf). New Methodology and Synthesis of Natural Product. The University of North Carolina at Chapel Hill. Дата обращения: 23 сентября 2009. Архивировано 23 апреля 2012 года.
- ↑ Ли Дж. Нозаки-Хияма-Киши (Nozaki-Hiyama-Kishi). Реакция // Именные реакции. Механизмы органических реакций = Name reactions / Пер. с англ. В.М.Демьянович. — М.: БИНОМ. Лаборатория знаний, 2006. — С. 249. — ISBN 5-94774-368-X.
- ↑ Kallemeyn J. M. The Nozaki-Hiyama-Kishi reaction (англ.) (pdf) (недоступная ссылка — история). The Department of Chemistry at the University of Illinois at Urbana-Champaign (12 апреля 2002). Дата обращения: 15 сентября 2009. Архивировано 23 апреля 2012 года.
- ↑ Catalytic, Nucleophilic Allylation of Aldehydes with Allyl Acetate (англ.). Organic Letters. ACS Publications. Дата обращения: 16 сентября 2009. Архивировано 23 апреля 2012 года.
- ↑ Pd-Catalyzed Nucleophilic Alkylation of Aliphatic Aldehydes with Allyl Alcohols: Allyl, 2-Tetrahydrofuryl, and 2-Tetrahydropyranyl Ethers as Useful C3, C4, and C5 Sources (англ.) (недоступная ссылка — история). Angewandte Chemie. Wiley InterScience. Дата обращения: 16 сентября 2009. Архивировано 23 апреля 2012 года.
- ↑ Nishiyama Y., Kakushou F., Sonoda N. Rhenium complex-catalyzed allylation of aldehydes with allyltributylstannane (англ.) // Tetrahedron Letters. — 2005. — Vol. 46, no. 5. — P. 787-789.
- ↑
Канниццаро реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 2. — С. 603-604. - ↑ 1 2 Глава 5.3.7. Реакция Канниццаро // Общая органическая химия. Кислородсодержащие соединения = Comprehensive Organic Chemistry / Под ред. Д.Бартона и В.Д.Оллиса. — М.: «Химия», 1982. — Т. 2. — С. 737-739.
- ↑
Mарч Дж. Глава 19. Реакции окисления и восстановления // Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: «Мир», 1988. — Т. 4. — С. 337-338. - ↑ Cannizzaro Reaction (англ.). Name Reactions. Organic Chemistry Portal. Дата обращения: 23 сентября 2009. Архивировано 23 апреля 2012 года.
- ↑ 1 2 3 4
Mарч Дж. Глава 16. Реакции присоединения к кратным связям углерод-гетероатом // Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: «Мир», 1988. — Т. 3. — С. 324-427. - ↑ 1 2 3
Альдольная конденсация // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 202-204. - ↑
Иванова реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 2. — С. 344—345. - ↑ Алкены II.Окислительное расщепление алкенов. www.chemnet.ru. Дата обращения: 24 февраля 2021. Архивировано 15 июня 2020 года.
План урока:
Формула и строение спиртов
Классификация спиртов
Номенклатура спиртов
Изомерия спиртов
Методы получения спиртов
Физические свойства
Химические свойства
Применение спиртов
Формула и строение спиртов
Спирты из-за наличия функциональной группы -ОН можно рассматривать как производное воды Н-О-Н. Геометрическое строение воды и спиртов схоже. Угол связи R-O-H равен 109˚, при этом гидроксильный кислород находится в состоянии sp3-гибридизации.

Строение молекулы спирта
У спиртов особенное электронное строение. Алкоголи – дипольные молекулы, которые содержат связи C-H, C-O, O-H. Атом кислорода имеет частично отрицательный заряд, а атомы углерода и водорода – частично положительный. Связь О-Н имеет большую полярность, по сравнению со связью С-О. Это явление связано с разностью электроотрицательности кислорода и водорода. Но полярность связей недостаточна для диссоциации и образования ионов Н+. Поэтому можно сделать вывод, что спирты – неэлектролиты.

Формула спиртов: CnH2n+1OH
Классификация спиртов
В классификации спиртов заключены особенности строения молекул. По числу гидроксильных групп различают одноатомные, двухатомные, трехатомные и многоатомные спирты.

Также спирты классифицируют в зависимости от положения гидроксильной группы на первичные, вторичные и третичные.

Номенклатура спиртов
Для спиртов свойственно несколько типов номенклатуры.
- Тривиальная (историческая) номенклатура. Для простых спиртовых соединений свойственны упрощенные названия. В этом случае название радикала переводят в прилагательное с помощью окончания «овый» и добавляют слово «спирт». Например, CH3-CH2-CH2-OH – пропановый спирт.
У первых двух представителей гомологического ряда есть особенные исторические названия. Метанол – древесный спирт, а этанол – винный. Такие названия обусловлены историческим методом получения. Опьяняющие свойства этанола были известны не менее чем за 8000 лет до н.э.
- Систематическая номенклатура спиртов. Как правило, в химии используют именно этот вид номенклатуры. В одноатомных спиртах к названию радикала добавляется суффикс «ол», в двухатомных – «диол», в трехатомных – «триол». Положение гидроксигруппы обозначается наименьшим значением, если в составе спирта отсутствует карбонильная и/или карбоксильная группа.

Алгоритм названия спиртов
Систематическая номенклатура подчиняется определенному алгоритму.
- Выбор главной цепи в соединении.
- Нумерация начинается с того конца, к которому ближе функциональная группа.
- Название углеводородного радикала.
- Прибавление окончания «ол» и указание номера атома углерода, с которым связана гидроксогруппа.

Изомерия спиртов
Для спиртов свойственно несколько видов изомерии – изомерия углеродного скелета, положения заместителя и межклассовая (с простыми эфирами). Изомерия углеродного скелета начинается с бутанола.

Типы изомерии спиртов
Способы получения спиртов
Существует несколько реакций получения спиртов.

Реакция проходит по правилу Марковникова, т.е. атом водорода присоединяется к более гидрированному атому углерода, а гидроксильная группа – к менее. Например, в молекуле CH3-CH2-CH=CH2 атом водорода Н+ примыкает к атому углерода, стоящему в СН2 у кратной связи, а гидроксильная группа ОН– – к СН.

Физические свойства
Физические свойства определяются особенностями строения молекулы спирта. Алкоголи – бесцветные жидкости с характерным запахом. Температуры плавления и кипения спиртов выше, чем у соответствующих представителей других классов веществ. По гомологическому ряду они увеличиваются.
Все алкоголи имеют плотность ниже единицы, т.е. они плавают на поверхности воды. Спирты растворимы в большинстве органических растворителях.
Особенность алкоголей заключается в том, что в гомологическом ряде нет газов. Агрегатное состояние спиртов – жидкое или твердое. Это связано с тем, что атом кислорода в гидроксильной группе обладает частично отрицательным зарядом, а атом водорода – частично положительным. Кислород притягивает положительно заряженные атомы и образует с ними водородные связи. Большое количество таких связей обеспечивает «прилипание» молекул спирта между собой и обуславливает особенное строение.

Из этилового спирта изготавливают алкогольные напитки. Несмотря на относительную безопасность употребления этанола, его систематическое употребление пагубно влияет на организм:
- 100 мл пива – гибель 3000 клеток головного мозга,
- 100 мл вина – гибель 5000 клеток головного мозга,
- 100 мл водки – гибель 7500 клеток головного мозга.
В алкогольные напитки вместо этанола могут добавлять метанол, который опасен для жизни. Эти два спирта отличаются по характеру пламени:
- метиловый горит зеленым пламенем,
- этиловый – синим.
Но при наличии примесей в метаноле, зеленое пламя может и не появиться.
Химические свойства
Реакции замещения
- Замещение водорода в гидроксильной группе
- Взаимодействие с активными металлами (например, с натрием)
Реакция проводится в безводной среде. В воде алкоголяты металлов полностью гидролизуются.
2CH3-CH2-O[H] + 2Na → 2CH3-CH2-ONa + H2
- Взаимодействие с кислотами (этерификация)
CH3-O[H + HO]-NO2→ CH3-O-NO2 + H2O
- Замещение гидроксигруппы
- Взаимодействие с галогеноводородами при нагревании
CH3-CH2-[OH + H]Br→ CH3-CH2-Br + H2O
- Взаимодействие с аммиаком
Реакция идет при пропускании смеси паров спирта с аммиаком при 300˚С над оксидом алюминия.
CH3-CH2-[OH +H]-NH2→CH3-CH2-NH2 + H2O
При избытке в спирте алкильных радикалов в молекуле аммиака могут замещаться два или три атома водорода.
2CH3-CH2-[OH + H]-NH2 → C2H5-NH-C2H5 + 2H2O
Реакции отщепления
- Дегидратация
- Межмолекулярная дегидратация в присутствии концентрированной серной кислоты и при 140˚С
CH3-CH2-O[H + НО]-СН2-СН3→С2Н5-О-С2Н5 + Н2О
CH3-O[H + HO]-CH2-CH3→ CH3-O-C2H5 + H2O
- Внутримолекулярная дегидратация в присутствии концентрированной серной кислоты и при 170˚С
OH]-CH2-CH2-[H →CH2=CH2 + H2O
Для метанола не характерна внутримолекулярная дегидратация. Реакция вторичных и третичных спиртов проходит по правилу Зайцева. Т.е. при отщеплении воды от спирта атом водорода отрывается от соседнего менее гидрированного атома углерода. Например, в молекуле CH3-CH2-CH(OH)-CH3 атом водорода Н+ отщепляется от атома углерода в СН3, находящегося вблизи гидроксильной группы.
CH3-CH(OH)-CH2-CH3→CH3-CH=CH-CH3 + H2O
- Дегидрирование в присутствии катализатора меди и под действием нагревания

Реакции окисления
- Полное окисление (горение)
CH3-CH2-OH + 3О2→ 2СО2 + 3Н2О
Неполное окисление

Качественная реакция на многоатомные спирты
Качественная реакция – химическое превращение, которое сопровождается характерными признаками. С ее помощью можно распознать определенное вещество.Строение многоатомных спиртов, т.е. присутствие в молекуле нескольких гидроксильных групп, обуславливает образование при взаимодействии со свежеполученным осадком гидроксида меди (II) растворимых в воде комплексов василькового цвета.

Строение одноатомных спиртов такая качественная реакция не подтверждает.
Применение спиртов
Метанол, или древесный спирт, опасен для употребления. Он был получен путем перегонки твердых древесных пород. Этот одноатомный спирт недобросовестные производители алкогольных напитков применяют вместо этанола, что провоцирует гибель потребителей. Древесный спирт в организме под влиянием фермента алкогольдегидрогеназы преобразуется в формальдегид и муравьиную кислоту, которые провоцируют слепоту. 50 мл метанола – смертельная доза. Метанол непросто отличить от этанола, т.к. они имеют схожий запах и вид.

Применение метилового спирта
Области применения этилового спирта разнообразны. Этиловый спирт используют в получении синтетического каучука, лекарственных препаратов и применяют как растворитель. Этанол используется в изготовлении алкоголя. При попадании в организм он снижает болевые ощущения, уменьшает торможения в коре головного мозга, ускоряет мочеобразование, провоцирует расширение кровеносных сосудов. При больших количествах этанола происходит его окисление до ацетальдегида, что вызывает тяжелые отравления.
При систематическом употреблении алкоголя снижается работоспособность головного мозга, а клетки печени погибают. Дети и подростки, употребляющие алкоголь, подвержены инфекционным заболеваниям. Продолжительность жизни пьющих людей меньше на 10-12 лет по сравнению с людьми, ведущих здоровый образ жизни.

Применение этилового спирта.
Практическое применение трехатомный спирт глицерин нашел в косметической, медицинской и пищевой промышленности. Он смягчает и успокаивает кожу. Также глицерин входит в состав зубной пасты, что предотвращает ее высыхание.

Применение глицерина
Многоатомный спирт глицерин также применяют в промышленной отрасли для предотвращения кристаллизации продуктов. Его используют как увлажнитель для табака. Он входит в состав клеев и предохраняет их от слишком быстрого высыхания.
Модель молекулы простейшего спирта — метанола
Спирты (от лат. spiritus — дух; устар. алкоголи, от араб. الكحول аль-кухуль — порошок) — органические соединения, содержащие одну или более гидроксильных групп (гидроксил, −OH), непосредственно связанных с насыщенным (находящимся в состоянии sp³-гибридизации) атомом углерода. Спирты можно рассматривать как производные воды (H−O−H), в которых один атом водорода замещен на органическую функциональную группу: R−O−H.
В номенклатуре ИЮПАК для соединений, в которых гидроксильная группа связана с ненасыщенным (sp²-гибридным) атомом углерода, рекомендуются названия «енолы» (гидроксил связан с винильной C=C-связью) и «фенолы» (гидроксил связан с бензольным или другим ароматическим циклом).
Спирты представляют собой обширный и разнообразный класс соединений: они весьма распространены в природе и часто выполняют важные функции в живых организмах. Спирты являются важными соединениями с точки зрения органического синтеза, не только представляя интерес как целевые продукты, но и как промежуточные вещества, имеющие ряд уникальных химических свойств. Кроме того, спирты являются промышленно важными продуктами и находят широчайшее применение как в промышленности, так и в повседневных приложениях.
Содержание
- 1 Этимология
- 2 Классификация спиртов
- 3 Номенклатура спиртов
- 3.1 Систематическая номенклатура
- 3.2 Другие номенклатуры
- 4 История открытия спиртов
- 5 Нахождение в природе
- 6 Метаболизм спиртов в организме человека
- 6.1 Физиологическая роль спиртов
- 6.2 Токсичность спиртов
- 7 Физические свойства и строение спиртов
- 8 Получение спиртов
- 8.1 Общие химические методы получения спиртов
- 8.2 Промышленные методы получения спиртов
- 9 Химические свойства спиртов
- 9.1 Диссоциация и кислотно-основные свойства спиртов
- 9.1.1 Кислотные свойства спиртов
- 9.1.2 Основные свойства спиртов
- 9.2 Нуклеофильное замещение
- 9.3 Дегидратация
- 9.4 Окисление
- 9.1 Диссоциация и кислотно-основные свойства спиртов
- 10 Идентификация спиртов
- 10.1 Химические методы идентификации спиртов
- 10.1.1 Качественный анализ гидроксильных групп
- 10.1.2 Количественный анализ спиртов
- 10.2 Спектральные методы анализа спиртов
- 10.2.1 Масс-спектрометрические методы анализа спиртов
- 10.2.2 ИК-спектроскопические методы анализа спиртов
- 10.2.3 ЯМР-спектроскопические методы анализа спиртов
- 10.1 Химические методы идентификации спиртов
- 11 Применение спиртов
- 11.1 Экономическое значение
- 11.2 Применение в органическом синтезе
- 11.3 Применение в качестве топлива
- 11.4 Промышленное применение спиртов
- 11.4.1 Растворители
- 11.4.2 Поверхностно-активные и моющие вещества
- 11.4.3 Полимерные материалы и их компоненты
- 11.4.4 Гидравлические жидкости и смазочные материалы
- 11.4.5 Пестициды
- 11.5 Применение спиртов в производстве потребительской продукции
- 11.5.1 Пищевая промышленность
- 11.5.2 Парфюмерия, косметика и бытовая химия
- 11.6 Применение спиртов в медицине
- 11.7 Прочие направления использования
- 12 Примечания
Этимология
Слово алкого́ль происходит от араб. الكحل (al-kuḥl) — «порошкообразная сурьма». Понятие берёт начало от алхимической методики восстановления химических веществ до «порошка», являющегося, как предполагалось, чистой эссенцией вещества. Подобным образом, и этиловый спирт является эссенцией вина, от чего и произошло его название. В русский язык слово пришло через нем. Alkohol, нидерл. alkohol или порт., исп. alcohol. Однако в русском языке сохранился в виде архаизма, по всей видимости, и омоним слова «алкоголь» в значении «мелкий порошок».
Слово спирт появилось в русском языке во времена Петра I через английское слово spirit, которое, в свою очередь, произошло от латинского spīritus — «дыхание, дух, душа».
Классификация спиртов
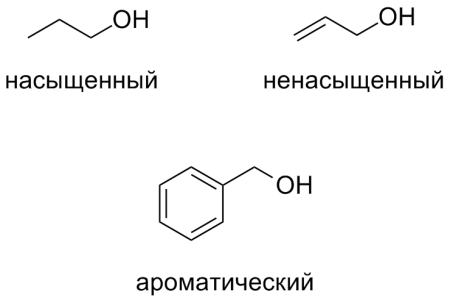
 |
 |
|
|
Примеры спиртов с различным числом гидроксильных групп |
Насыщенный, ненасыщенный и ароматический спирты |
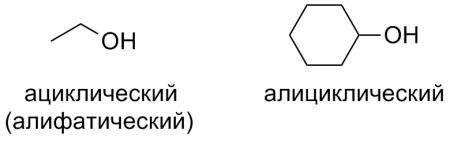 |
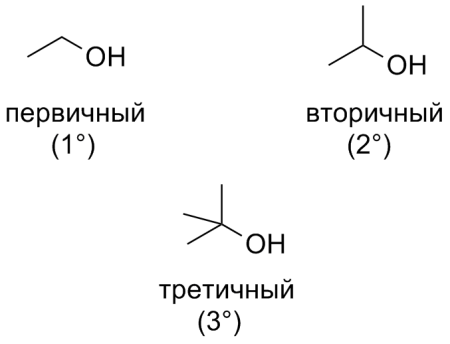 |
|
|
Ациклический и циклический спирты |
Первичный, вторичный и третичный спирты |
Спирты классифицируются следующим образом (в скобках приведены примеры):
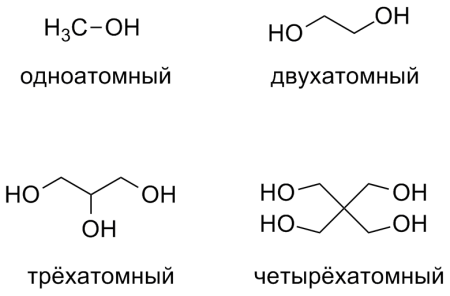
- По числу гидроксильных групп:
— одноатомные спирты (метанол);
— двухатомные спирты (этиленгликоль);
— трёхатомные спирты (глицерин);
— четырёхатомные спирты (пентаэритрит);
— многоатомные спирты (пятиатомный спирт: ксилит).
- В зависимости от насыщенности углеводородного заместителя:
— предельные (насыщенные) спирты (бутанол);
— непредельные (ненасыщенные) спирты (аллиловый спирт, пропаргиловый спирт);
— ароматические спирты (бензиловый спирт).
- В зависимости от наличия или отсутствия цикла в углеводородном заместителе:
— ациклические (алифатические) спирты (этанол);
— алициклические спирты (циклогексанол).
- В зависимости от числа заместителей при α-углеродном атоме:
— первичные спирты (этанол);
— вторичные спирты (пропанол-2);
— третичные спирты (2-метилпропанол-2).
Номенклатура спиртов
Систематическая номенклатура
Основная статья: Систематическая номенклатура спиртов и фенолов
По номенклатуре ИЮПАК названия простых спиртов образуются от названий соответствующих алканов с добавлением суффикса «-ол», положение которого указывается арабской цифрой.
Правила построения названий спиртов:
1. Выбирают родительский углеводород по самой длинной непрерывной углеводородной цепи, содержащей гидроксильную группу. Он формирует базовое название (по числу атомов углерода).
2. Родительский углеводород нумеруют в таком направлении, чтобы гидроксильная группа получила наименьший номер в названии. (Если в соединении имеются функциональные группы старше гидроксильной, то это правило применяется к старшей функциональной группе.)
3. Старшая функциональная группа обозначается в виде суффикса (для гидроксильной — -ол), а остальные заместители — в виде приставок в алфавитном порядке. Их положение в углеводородной цепи обозначается при помощи цифр — локантов, помещаемых после суффиксов и перед приставками. Для многоатомных спиртов перед суффиксом -ол указывается число гидроксильных групп (-диол, -триол, -тетраол и т. д.).
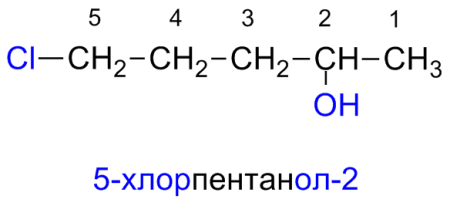
4. Если при различных вариантах нумерации цепи гидроксильная группа получает один и тот же локант, то цепь нумеруют в том направлении, при котором другой заместитель получает наименьший локант.
Другие номенклатуры
- Радикало-функциональная номенклатура. В рамках данной номенклатуры название образуется от названия класса соединения (спирт) с добавлением названий радикалов, присоединённых к гидроксильной группе, например: этиловый спирт C2H5OH, гексиловый спирт C6H13OH, аллиловый спирт СH2=CH-CH2OH.
- Рациональная номенклатура (карбинольная номенклатура) спиртов рассматривает их как производные метанола CH3OH, называемого в данном случае карбинолом: диметилкарбинол (СH3)2CHOH, трифенилкарбинол (C6H5)3COH.
- Тривиальная номенклатура. В популярной и научной литературе можно нередко встретить исторические, или тривиальные, названия спиртов, которые вследствие сложившейся традиции используются вместо систематических названий. Тривиальные названия обычно происходят от названия природного источника получения того или иного спирта. Так, например, метанол называют древесным спиртом, этанол — винным спиртом, гераниол содержится в гераниевом масле, а цетиловый спирт ранее получали из жира кашалотов (лат. cetus — кит). Некоторые спирты получили тривиальные названия из-за своих физических свойств (глицерин от др.-греч. glykeros, сладкий). Спирты, производные от природных углеводов, сохраняют корень тривиального названия соответствующих углеводов (маннит, ксилит, сорбит).
История открытия спиртов
Хмельной растительный напиток, содержащий этанол, был известен человечеству с глубокой древности. Считается, что не менее чем за 8000 лет до нашей эры люди были знакомы с действием перебродивших фруктов, а позже — с помощью брожения получали хмельные напитки, содержащие этанол, из фруктов и мёда. Археологические находки свидетельствуют, что в Западной Азии виноделие существовало ещё в 5400—5000 годах до н. э., а на территории современного Китая, провинция Хэнань, найдены свидетельства производства ферментированных смесей из риса, мёда, винограда и, возможно, других фруктов, в эпоху раннего неолита: от 6500 до 7000 гг. до н. э.
Впервые спирт из вина получили в VI—VII веках арабские химики; способ получения спирта содержится в записях персидского алхимика Ар-Рази. В Европе этиловый спирт был получен из продуктов брожения в XI—XII веке, в Италии.
В Россию спирт впервые попал в 1386 году, когда генуэзское посольство привезло его с собой под названием «аква вита» и презентовало великокняжескому двору.
В 1661 году английский химик Роберт Бойль впервые получил метанол перегонкой древесины. Абсолютированный этанол из его водного раствора был впервые получен в 1796 году русским химиком Т. Е. Ловицем при перегонке над поташом.
В 1842 году немецкий химик Я. Г. Шиль открыл, что спирты образуют гомологический ряд, отличаясь на некоторую постоянную величину. Однако, он ошибся, описав её как C2H2. Спустя два года, другой химик Шарль Жерар установил верную гомологическую разницу CH2 и предсказал формулу и свойства неизвестного в те годы пропилового спирта. В 1850 году английский химик Александр Вильямсон, исследуя реакцию алкоголятов с этилиодидом, установил, что этиловый спирт является производным воды с одним замещённым атомом водорода, экспериментально подтвердив формулу C2H5OH. Впервые синтез этанола действием серной кислоты на этилен осуществил в 1854 году французский химик Марселен Бертло.
Первое исследование метилового спирта было сделано в 1834 году французскими химиками Жаном-Батистом Дюма и Эженом Пелиго. Они назвали его «метиловым или древесным спиртом», так как он был обнаружен в продуктах сухой перегонки древесины. Синтез метанола из метилхлорида осуществил французский химик Марселен Бертло в 1857 году. Им же в 1855 году был открыт изопропиловый спирт, полученный действием серной кислоты на пропилен.
Впервые третичный спирт (2-метилпропанол-2) синтезировал в 1863 году известный русский химик А. М. Бутлеров, положив начало целой серии экспериментов в этом направлении.
Двухатомный спирт этиленгликоль впервые был синтезирован французским химиком А. Вюрцем в 1856 году. Глицерин был обнаружен в природных жирах ещё в 1783 году шведским химиком Карлом Шееле, однако его состав был открыт только в 1836 году, а синтез осуществлен из ацетона в 1873 году Шарлем Фриделем.
Нахождение в природе
Природный спирт ментол встречается в мяте и герани
Спирты широко распространены в природе как в свободном виде, так и в составе сложных эфиров.
Метиловый спирт в небольшом количестве содержится в некоторых растениях, например: борщевике (Heracleum).
Этиловый спирт является естественным продуктом анаэробного брожения органических продуктов, содержащих углеводы, под действием дрожжей рода Saccharomyces и бактерий Zimomonas и часто образуется в прокисших ягодах и фруктах. При этом углеводы через последовательность ферментативных реакций, именуемую гликолизом, превращаются в пируват, который далее под действием пируватдекарбоксилазы переходит в ацетальдегид. Последний затем в присутствии алкогольдегидрогеназы акцептирует восстановительный эквивалент от восстановленной формы NAD (восстановление NAD происходит на одной из стадий гликолиза), что даёт этанол в качестве конечного продукта. Данный процесс не только является причиной накопления этанола в природных объектах, но и активно используется в виноделии, пивоварении и хлебопекарной промышленности. В ходе брожения могут образовываться также и другие спирты, например, изопропанол, бутанол-1, бутандиол-2,3.
В эфирных маслах зелёных частей многих растений содержится (Z)-гексен-3-ол-1 («спирт листьев»), придающий им характерный запах. Также в растительном мире очень широко представлены терпеновые спирты, многие из которых являются душистыми веществами, например: борнеол (компонент древесины борнеокамфорного дерева), ментол (в мяте, герани), гераниол и цитронеллол (компоненты цветочных эфирных масел), фенхол (в смоле хвойных деревьев и плодах фенхеля) и др.
В животном и растительном мире распространены конденсированные тетрациклические спирты (производные гонана), обладающие высокой биологической активностью и входящие в класс стероидов, например, холестерин, содержащийся в клетках практически всех живых организмов, особенно животных. Отдельную группу стероидов составляют жёлчные многоатомные спирты, находящиеся в жёлчи животных и человека: буфол, холестантетрол, холестанпентол, миксинол, сцимнол, химерол и др.
В природе встречаются разнообразные спиртовые производные углеводов, например, сорбит (содержится в ягодах вишни и рябины) маннит (в ясене) и др.
В 1959 году немецким химиком Адольфом Бутенандтом при изучении экстракта самки тутового шелкопряда был открыт половой аттрактант, названный бомбиколом. Дальнейшее изучение феромонов насекомых показало, что значительная часть этих феромонов представлена спиртами.
Метаболизм спиртов в организме человека
Физиологическая роль спиртов
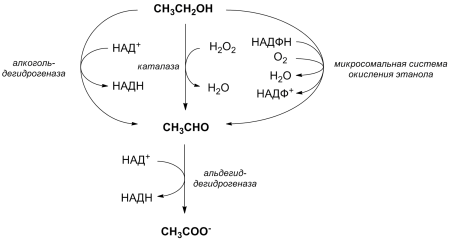
Метаболизм этанола в организме человека
Многие спирты являются участниками важных биохимических процессов, происходящих в живом организме. Так, некоторые витамины относятся к классу спиртов, например, витамин А (ретинол), витамин D (эргокальциферол и др.). Стероидные гормоны, среди которых имеются и спирты (эстрадиол, кортизол и др.), участвуют в регуляции обмена веществ и некоторых физиологических функциях организма.
Глицерин является основой более чем половины природных липидов, которые представляют собой его сложные эфиры с жирными кислотами и являются источниками энергии для организма. Также глицерин участвует в глюконеогенезе — процессе образования глюкозы в печени. При этом глицерин под действием ферментов превращается в глицеральдегид-3-фосфат, который далее попадает в метаболический путь глюконеогенеза. Физиологически важным полиолом является мио-инозитол.
Среди низших спиртов с точки зрения физиологии наибольший интерес представляет, несомненно, этанол. В организме человека этанол является естественным метаболитом и в норме присутствует в крови в очень низких концентрациях. Также этанол может поступать в организм с пищей. Этанол в организме человека метаболизируется преимущественно в печени. Под действием цитозольного фермента алкогольдегидрогеназы этанол окисляется в ацетальдегид, который далее перерабатывается митохондриальной альдегиддегидрогеназой в ацетат. Ацетат после активации короткоцепочечной ацил-коэнзим А-синтетазой может далее разрушаться в цикле Кребса. В утилизации этанола второстепенную роль играет также микросомальная этанол-окисляющая система, представленная цитохромом P450 и каталазой. При высокой концентрации алкоголя в крови ферменты не справляются с оксилением ацетальдегида до ацетата, и в организме происходит накопление ацетальдегида, который в 10—30 раз токсичнее этанола, за счёт чего происходит отравление организма, т. н. похмелье. По энергетической ценности для организма этанол (7 ккал/г) занимает промежуточное положение между углеводами (4,1 ккал/г) и жирами (9,3 ккал/г). Вклад этанола в общую калорийность пищи у не страдающих алкоголизмом взрослых людей может достигать 12 %. Однако потребление этанола в качестве пищевого продукта и источника энергии имеет ряд недостатков с биохимической точки зрения. Кроме образования токсичного ацетальдегида, к таким недостаткам следует причислить тот факт, что избыточные калории, поступившие в организм в форме этанола, могут запасаться только в жирах, так как возможность преобразования этанола в углеводы в организме человека отсутствует. Кроме того, этанол нарушает другие метаболические процессы: ингибирует глюконеогенез (это является причиной гипогликемии при приёме больших доз алкоголя), ускоряет производство цитокинов, изменяет концентрацию гормонов. Алкогольные напитки содержат очень мало витаминов и минеральных веществ, что также может оказать негативное влияние на здоровье. Следует также отметить, что пищевой этанол сам по себе намного дороже, чем энергетически эквивалентное количество сахара.
Токсичность спиртов
См. также: Токсикология этанола
Одноатомные предельные спирты вводят организм в наркозоподобное или гипнотическое состояние, а также оказывают токсическое действие.
Метиловый спирт — сильный яд (особенно при приёме внутрь) нервного и сердечно-сосудистого действия с выраженным кумулятивным эффектом; поражает органы зрения вплоть до полной слепоты. В больших дозах (30 г и более) вызывает смерть.
Этиловый спирт обладает токсическим эффектом. Быстро всасывается через слизистую оболочку желудка и тонкого кишечника, достигая максимальной концентрации в крови через 60—90 минут после его приёма. Этанол вызывает сначала возбуждение, а затем резкое угнетение центральной нервной системы (в том числе разрушает мозговую оболочку); его употребление приводит к нарушению важнейших функций организма, тяжелому поражению органов и систем. Оказывает эмбриотоксическое и тератогенное действие.
Изопропиловый спирт по своему токсическому воздействию напоминает этанол, вызывая угнетение центральной нервной системы и поражая внутренние органы. В высокой концентрации приводит к коме, конвульсиям и летальному исходу (около 3—4 г/кг).
В связи с широким использованием простейших спиртов в различных отраслях промышленности и, в частности, в качестве растворителей, опасным является их ингаляционное воздействие. Острое токсичное воздействие спиртов, испытанное на крысах, проявилось в следующих ингаляционных концентрациях:
- метиловый спирт: 3,16 % в течение 18—21 часов — 100 % летальность; 2,25 % в течение 8 часов — наркотический эффект; 0,8 % в течение 8 часов — летаргия;
- этиловый спирт: 3,2 % в течение 8 часов — частичная летальность; 2,2 % в течение 8 часов — глубокий наркоз; 0,64 % в течение 8 часов — летаргия;
- изопропиловый спирт: 1,2 % в течение 8 часов — 50 % летальность; 1,2 % в течение 4 часов — наркотический эффект.
Этиленгликоль очень токсичен при пероральном попадании в организм, поражает ЦНС и почки. Смертельная доза составляет 1,4 г/кг массы тела.
Физические свойства и строение спиртов

Геометрия связи C−O−H в молекуле метанола
Пространственное строение метанола
Молекулы спиртов, подобно молекуле воды, имеют угловое строение. Угол R−O−H в молекуле метанола равен 108,5°. Атом кислорода гидроксильной группы находится в состоянии sp³-гибридизации. Спирты имеют существенно более высокие температуры плавления и кипения, чем можно было бы предполагать на основании физических свойств родственных соединений. Так, из ряда монозамещённых производных метана, метанол имеет необычно высокую температуру кипения, несмотря на относительно небольшую молекулярную массу:
Молекулярные массы и температуры кипения метана и некоторых его производных
| Метан CH4 |
Метанол CH3OH |
Хлорметан CH3Cl |
Нитрометан CH3NO2 |
Бромметан CH3Br |
|
|---|---|---|---|---|---|
| Молярная масса, г/моль | 16,04 | 32,04 | 50,48 | 61,04 | 94,94 |
| Температура кипения, °С | −161,5 | 64,5 | −24,2 | 101,2 | 3,6 |
Высокие температуры кипения спиртов объясняются наличием межмолекулярных водородных связей. Энергия водородной связи значительно ниже, чем энергия ковалентной химической связи. Так, например, для метанола энергия водородной связи составляет 16,7 кДж/моль , тогда как связи C-H, C-O и O-H имеют энергию 391,7, 383,5 и 428,8 кДж/моль соответственно. Тем не менее, влияние водородных связей на физические свойства спиртов весьма значительное.
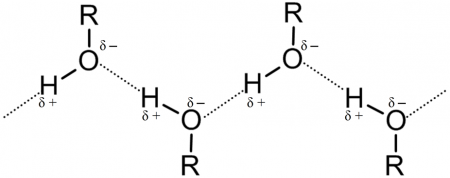
Межмолекулярные водородные связи в спиртах
Молекулы спирта, имея две полярных связи C−O и O−H, обладают дипольным моментом (~5,3—6,0⋅10−30 Кл·м). Электростатические заряды в молекуле метанола составляют: на атоме углерода 0,297 e; на атоме гидроксильного водорода 0,431 e; на атоме кислорода −0,728 e. Вместе с тем, энергия ионизации спиртов ниже, чем у воды (10,88 эВ для метанола против 12,61 эВ для воды), что объясняется электронодонорным эффектом алкильной группы.
Следует отметить, что влияние гидроксильной группы особенно велико на соединения с небольшой углеводородной цепочкой. Так, например, метанол и этанол неограниченно смешиваются с водой и имеют довольно высокие плотности и температуры кипения для своей молекулярной массы, в то время как высшие спирты гидрофобны и мало отличаются по свойствам от соответствующих углеводородов.
Некоторые физические константы алифатических предельных спиртов
| Название | Формула | Т. кип., °С | Т. пл., °С | Плотность, кг/м3 (20 °С) | Показатель преломления, nD20 |
|---|---|---|---|---|---|
| Метанол | CH3OH | 64,7 | −97,78 | 791,5 | 1,32855 |
| Этанол | C2H5OH | 78,3 | −114,65 | 789,5 | 1,36139 |
| Пропанол-1 | C3H7OH | 97,2 | −124,10 | 803,5 | 1,38556 |
| Пропанол-2 | CH3CH(CH3)OH | 82,5 | −87,95 | 786,2 | 1,37711 |
| Бутанол-1 | C4H9OH | 117,8 | −88,64 | 808,6 | 1,39929 |
| 2-Метилпропанол-1 | (CH3)2CHCH2OH | 108,0 | −101,97 | 802,1 | 1,39549 |
| Бутанол-2 | CH3CH2CH(ОН)CH3 | 99,5 | −114,70 | 806,0 | 1,39240 |
| 2-Метилпропанол-2 | (CH3)2C(OH)CH3 | 82,9 | 25,82 | 765,2 | 1,38779 |
| Пентанол-1 | C5H11OH | 138,0 | −77,59 | 813,3 | 1,40999 |
| Гексанол | C6H13OH | 157,1 | -47,40 | 821,7 | 1,41816 |
| Гептанол | C7H15OH | 176,3 | −32,80 | 824,0 | 1,42351 |
| Октанол | C8H17OH | 195,1 | −16,30 | 822,7 | 1,42920 |
| Нонанол | C9H19OH | 213,5 | −5,00 | 827,0 | 1,43325 |
| Деканол | C10H21OH | 231,0 | 6,00 | 826,0 | 1,43660 |
Некоторые физические константы алициклических, ароматических и непредельных спиртов
| Название | Формула | Т. кип., °С | Т. пл., °С | Плотность, кг/м3 (20 °С) | Показатель преломления, nD20 |
|---|---|---|---|---|---|
| Пропен-2-ол-1 | CH2=CHCH2OH | 96,9 | −129 | 852,0 | 1,4133 |
| Пропин-2-ол-1 | CH≡CCH2OH | 113,6 | −48 | 948,5 | 1,4322 |
| Циклогексанол | C6H11OH | 161,1 | 25,15 | 941,6 | 1,4648 |
| Фенилкарбинол | C6H5CH2OH | 205,0 | −15,3 | 1041,9 | 1,5396 |
| 2-Фенилэтанол | C6H5CH2CH2OH | 218,2 | −27,0 | 1020,2 | 1,5325 |
| 3-Фенилпропен-2-ол-1 | C6H5CH=CHCH2OH | 256—258 | 34 | 1044,0 | 1,5819 |
| 2-Фурилкарбинол | (C4H3O)CH2OH | 155 | — | 1131,9 | 1,5324 |
Некоторые физические константы многоатомных спиртов
| Название | Формула | Т. кип., °С | Т. пл., °С | Плотность, кг/м3 (20 °С) | Показатель преломления, nD20 |
|---|---|---|---|---|---|
| Глицерин | HOCH2CH(OH)CH2OH | 290 | 20 | 1260,4 | 1,4729 |
| Пентаэритрит | C (CH2OH)4 | 263,5 | 1397 | 1,548 | |
| Этиленгликоль | OHCH2CH2OH | 197,85 | –12,6 | 1,1155 | 1,432 |
Получение спиртов
Основная статья: Получение спиртов
Общие химические методы получения спиртов
Спирты могут быть получены из самых разных классов соединений, таких как углеводороды, галогеналканы, амины, карбонильные соединения, эпоксиды. В основном, все методы сводятся к реакциям окисления, восстановления, присоединения и замещения.
Спирты получают, окисляя алканы и циклоалканы под действием сильных неорганических окислителей: озона, перманганата калия, оксида хрома (VI), хромовой кислоты, диоксида селена, пероксида водорода, а также некоторых надкислот. Из-за возможности дальнейшего окисления получаемых спиртов, метод имеет значение только для получения третичных спиртов.
Окисление алкенов значительно более распространено в лабораторной практике, особенно для получения двухатомных спиртов — диолов. В зависимости от выбора реагента окисление можно провести с различной стереоселективностью: при действии на алкены тетраоксида осмия, перманганата калия, хлората натрия, йода с карбоксилатом серебра протекает син-гидроксилирование; для проведения анти-гидроксилирования используют пероксид водорода и надкислоты, оксиды молибдена (VI) и вольфрама (VI), оксид селена (IV) и пр.
Спирты образуются также при восстановлении альдегидов или кетонов под действием борогидрида натрия в протонном растворителе, а также алюмогидрида лития. Восстановление сложных эфиров и карбоновых кислот также производится под действием комплексных гидридов, обычно, алюмогидрида лития и приводит к спиртам.
Кислотно-катализируемое присоединение воды к алкенам приводит к образованию спиртов. В соответствии с правилом Марковникова, в данной реакции образуются более замещённые спирты. В лабораторной практике чаще используют аналогичную, но более мягкую реакцию оксимеркурирования — демеркурирования, а также реакцию гидроборирования — окисления, приводящую к продуктам, не согласующимся с правилом Марковникова.
Реакции нуклеофильного присоединения металлорганических соединений (ацетиленидов, реактивов Гриньяра, медь- и литийорганических соединений и т. д.) к карбонильным соединениям также приводят к спиртам, причём если присоединение происходит к формальдегиду HCHO, то образуются первичные спирты, если к другим альдегидам, то образуются вторичные спирты. Присоединение к кетонам даёт третичные спирты. Третичные спирты можно получить также путём присоединения двух эквивалентов металлорганического соединения к сложным эфирам.
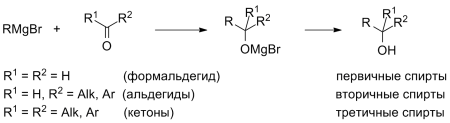
Спирты можно получать при обработке галогеналканов раствором щёлочи. Реакция протекает как нуклеофильное замещение и сопровождается рацемизацией (при мономолекулярном механизме) или обращением конфигурации (при бимолекулярном механизме). Важным препаративным методом является окисление алкилгалогенидов надпероксидом калия.
Промышленные методы получения спиртов
В промышленности спирты получают при помощи химических методов либо биохимических методов производства.
Единственным промышленно важным методом синтеза метанола является каталитическая реакция между оксидом углерода (II) и водородом. Сырьём в производстве метанола служит природный газ, который на первой стадии процесса подвергают очистке от соединений серы (сера является ядом для катализаторов, используемых на следующей стадии). Далее происходит паровая конверсия природного газа в синтез-газ (смесь СО и водорода), который после конденсации паров воды превращают в метанол на смешанном медно-цинко-хромовом катализаторе при температуре 250 °С и давлении до 10 МПа. Получаемый таким образом метанол содержит воду и примеси других спиртов (этанола, пропанола и более высших) и может быть очищен ректификацией. Мировое потребление метанола в 2015 году составило порядка 70 млн тонн.
Этанол и пропанол-2 получают методом гидратации соответствующих алкенов — этилена и пропилена. В промышленности используют два варианта гидратации: сернокислотную и каталитическую. Сернокислотная гидратация включает в себя абсорбцию этилена концентрированной серной кислотой (94—98 %) при температуре 80 °С и давлении 1,3—1,5 МПа и последующий гидролиз образующихся сульфоэфиров водой. Второй метод гидратации основан на использовании фосфорной кислоты, нанесённой на силикагель или другую подложку, в качестве катализатора. Смесь деионизированной воды и этилена нагревают до температуры 300 °С под давлением 6—8 МПа, а полученный этанол очищают ректификацией. Данные методы позволяют получить этанол, содержащий 5 % воды по массе. Получение безводного этанола (99,9 %) основано на азеотропном удалении воды с бензолом. По данным на 2003 год, мировое производство этанола только гидратацией этилена составляет 6 млн тонн в год.
Мировое производство топливного этанола в 2015 году по данным Renewable Fuel Association составило около 77 млн тонн.
Для гидратации пропилена требуются более мягкие условия. Сернокислотный процесс проводят при комнатной температуре и концентрации серной кислоты, равной 70—75 %, а каталитическая гидратация протекает при 180 °С и 4 МПа. Иногда для гидратации пропилена используют вольфрамовый катализатор (WO3·SiO2, 250 °C и 25 МПа)). Мировое производство пропанола-2 в 2008 году составляло 1,8 млн тонн.
Также реакцией гидратации в промышленности получают этиленгликоль — один из важнейших продуктов химической промышленности, производимый в количестве 19,9 млн тонн ежегодно и используемый в больших количествах для производства антифриза и волокон. Сырьём для данного синтеза служит окись этилена, получаемая прямым окислением этилена кислородом воздуха. Превращение окиси этилена в этиленгликоль происходит при нагревании её смеси с 20-кратным мольным избытком воды до 200 °С без катализатора. Этиленгликоль затем отделяется от воды и продуктов олигомеризации в результате последовательных ректификаций.
Промышленное получение пропанола-1 основано на реакции гидроформилирования этилена и последующем гидрировании полученного пропаналя. Гидроформилирование проводится при температуре 90—130 °С, общем давлении пропилена, оксида углерода(II) и водорода, равном 2,8 МПа, и в присутствии 500 мд родиевого катализатора. Вторая стадия протекает при 110—150 °С в избытке водорода на различных металлических катализаторах (используются комбинации соединений меди, цинка, никеля и хрома). Объёмы мирового производства пропанола-1 на 2003 год составили 0,14 млн тонн.
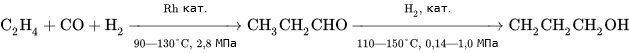
Глицерин получают как побочный продукт превращения жиров в жирные кислоты и метиловые эфиры жирных кислот. Данный процесс лежит в основе получения биодизеля, при этом на каждую тонну биодизеля образуется 100 кг глицерина. Таким методом синтезируют 1,5 млн тонн глицерина ежегодно (2004 год), причём по прогнозам эти объёмы будут расти в связи с увеличением интереса к альтернативным видам топлива. Жиры гидролизуют при 220—260 °С и давлении 2—6 МПа либо переэтерифицируют метанолом. Расщепление жиров под действием щёлочи либо карбонатов применяется в ограниченном масштабе, например, при производстве мыла.
Высшие жирные спирты производят несколькими способами, среди которых гидрогенолиз метиловых эфиров жирных кислот, получаемых переэтерификацией жиров, а также гидроформилирование алкенов и олигомеризация этилена с последующим окислением (метод Циглера). Ежегодно в мире производится 2,15 млн тонн высших жирных спиртов (2003 год).
Для некоторых спиртов более важную роль в промышленном синтезе играют биохимические методы. В частности, объёмы производимого биоэтанола значительно превышают объёмы синтетического этанола. В основе биохимического получения этанола лежит кислотный или ферментативный гидролиз растительного сырья с последующей анаэробной спиртовой ферментацией (сбраживанием) образующихся углеводов дрожжевыми грибами (Saccharomyces) или некоторыми видами бактерий. В частности, дрожжи являются весьма удобными микроорганизмами для широкого промышленного использования. Ферментация под действием дрожжей характеризуется высокой селективностью, низким накоплением побочных продуктов, высоким выходом этанола, высокой скоростью процесса, хорошей толерантностью дрожжей к повышенным концентрациям этанола и субстрата. Сырьём в данном процессе могут служить легко ферментируемые углеводы, а также крахмал и другие органические субстраты, которые необходимо предварительно гидролизовать до ферментируемой формы. Обычно используют сельскохозяйственные культуры (сахарная свёкла, картофель, зерновые культуры), продукты лесного хозяйства (древесина, солома) либо сельскохозяйственные отходы.
Производство биобутанола основано на ферментации углеводного сырья с использованием бактерий Clostridium acetobutylicum.
Химические свойства спиртов
Основная статья: Химические свойства спиртов
Химические свойства спиртов определяются наличием в них гидроксильной группы. Поскольку гидроксильная группа является полярной, она может гетеролитически диссоциировать, особенно, под действием сильных оснований. Таким образом, спирты проявляют свойства слабых кислот. Кроме того, высокая электроотрицательность кислорода обуславливает наличие электрофильного атома углерода и, соответственно, способность спиртов подвергаться реакциям нуклеофильного замещения под действием ряда нуклеофилов. Наконец, атом кислорода гидроксильной группы имеет нуклеофильный характер, поэтому спирты могут выступать нуклеофилами в реакциях замещения и присоединения. Также для спиртов характерны реакции окисления.
Диссоциация и кислотно-основные свойства спиртов
Кислотные свойства спиртов
Спирты способны проявлять как кислотные, так и основные свойства. Как слабые кислоты, спирты диссоциируют по связи O-H с образованием алкоксид-иона. Кислотные характеристики спиртов оценивают по константе кислотности Ka.
ROH + H2O ⇌ RO− + H3O+
pKa = −log Ka. В водном растворе кислотность спиртов снижается с увеличением молекулярной массы и разветвлённости углеводородной цепи. Это связывают с увеличением положительного индуктивного эффекта алкильных заместителей в данном ряду и уменьшением устойчивости образующегося алкоксид-иона за счёт локализации отрицательного заряда на атоме кислорода. В целом, электроноакцепторные заместители (-NO2, -CN, -F, -Cl, -Br, -I, -OR и др.) увеличивают кислотность спиртов (уменьшают pKa). Напротив, электронодонорные заместители (например, алкильные заместители) уменьшают кислотность спиртов (увеличивают pKa). Так, pKa 2,2,2-трифторэтанола имеет значение 12,43 (против 15,9 у этанола), а полностью фторированного трет-бутанола — 5,4 (против 17,7 у трет-бутанола). Сравнительная кислотность спиртов и соединений других классов схематически представлена на рисунке.
Как слабые кислоты, спирты вступают в реакции с щелочными, щелочноземельными и некоторыми другими металлами, и с сильными основаниями, например, гидридами или амидами металлов, реактивами Гриньяра.
Основные свойства спиртов
Спирты могут также вести себя как слабые основания Льюиса, образовывая с сильными минеральными кислотами соли алкоксония, а также давая донорно-акцепторные комплексы с кислотами Льюиса. Обычно подобные реакции не останавливаются на указанной стадии и ведут к нуклеофильному замещению гидроксильной группы или отщеплению воды.
ROH + HX ⇌ ROH2+ + X−
ROH + AlCl3 ⇌ ROH+ AlCl3− Количественно основность спиртов оценивают по константе основности pKb или связанной с ней константе кислотности сопряжённой кислоты pKaH+:
ROH2+ ⇌ ROH + H+
pKaH+ = −log KaH+. Спирты являются слабыми основаниями, и их основность возрастает с увеличением длины или разветвлённости углеводородного радикала при гидроксильной группе. Данный эффект наблюдается из-за роста положительного индуктивного эффекта радикала в данном ряду, за счёт которого увеличивается отрицательный заряд на атоме кислорода гидроксильной группы.
Нуклеофильное замещение
Основная статья: Реакции нуклеофильного замещения
Атом углерода, непосредственно соединённый с гидроксильной группой, имеет частичный положительный заряд, что делает возможной атаку нуклеофильной частицы (галогенид-иона, аммиака, спирта и др.) по этому атому углерода с замещением гидроксильной группы на эту частицу. Гидроксильная группа является плохой уходящей группой, поэтому обычно необходима её дополнительная активация.
- Реакции нуклеофильного замещения в спиртах легче протекают в кислой среде, поскольку гидроксильная группа спирта протонируется, и фактической уходящей частицей является не гидроксид-ион OH−, а молекула воды H2O. Кислотными свойствами может обладать сам реагент (часто используют галогеноводородные кислоты), так и специально добавленная неорганическая кислота, например, серная кислота.
- Замещение гидроксильной группы также протекает под действием галогенидов серы и фосфора (SOCl2, PBr3, PBr5, POCl3 и др.). В данном случае ключевую роль выполняет соединение серы или фосфора, образующее активированный интермедиат с молекулой спирта.
- Гидроксильную группу также превращают в сульфонатную группу, которая является хорошей уходящей группой. Для этих целей спирт сначала превращают в сульфонат, который затем подвергают реакции нуклеофильного замещения. В качестве реагентов для модификации гидроксильной группы обычно используют метансульфонилхлорид или п-толуолсульфонилхлорид (англ.)русск..
Дегидратация
В присутствии кислотных катализаторов (оксид алюминия, серная кислота, фосфорная кислота и др.) спирты могут подвергаться дегидратации с образованием алкенов. Например, дегидратация этилового спирта приводит к образованию этилена. Реакция протекает в соответствии с правилом Зайцева, согласно которому при дегидратации образуется более устойчивый, более замещённый при двойной связи алкен.
Окисление
- Под действием различных окислителей первичные спирты окисляются до альдегидов и далее — до карбоновых кислот, причём остановить реакцию на стадии образования альдегидов, предотвратив их дальнейшее окисление удаётся только за счёт использования специальных реагентов (хлорхромата пиридиния PCC и дихромата пиридиния PDC).
- Вторичные спирты окисляются до кетонов. Реакцию обычно проводят под действием реагента Джонса (CrO3—серная кислота). Дальнейшее окисление кетонов протекает только в жёстких условиях с разрушением углеродного скелета.
- Третичные спирты окисляются только в весьма жёстких условиях с разрушением углеродного скелета.
Идентификация спиртов
Химические методы идентификации спиртов
Качественный анализ гидроксильных групп

Проба Лукаса для этанола (слева) и трет-бутилового спирта (справа)
Наличие гидроксильной группы в соединении можно выявить несколькими распространёнными химическими реакциями.
- Проба Лукаса заключается в действии на спирт смеси соляной кислоты и хлорида цинка. При этом происходит образование алкилхлорида, который сначала образует эмульсию со спиртом, а затем отслаивается в виде второй фазы. Проба позволяет различить спирты с разным строением углеродной цепи: третичные спирты реагируют практически мгновенно, вторичные — примерно через 5 минут, а первичные реагируют очень медленно. Некоторые первичные спирты, активные в реакциях нуклеофильного замещения (аллиловый, бензиловый), также дают положительную реакцию с реактивом Лукаса.
- Йодоформная проба предназначена для идентификации метилкетонов и метилкарбинолов (RCH (OH)СH3) по реакции с йодом в щелочной среде. При этом происходит образование желтоватого осадка иодоформа, имеющего характерный запах.
- Проба Мейера позволяет дифференцировать первичные, вторичные и третичные спирты по реакции получаемых из них нитропроизводных с азотистой кислотой. На первой стадии спирты превращают в галогенопроизводные, а затем — в нитроалканы. При взаимодействии нитросоединений с HNO2 раствор приобретает красную окраску при подщелачивании, если исходный спирт был первичным; раствор в хлороформе становится синим, если спирт был вторичным. Третичные спирты дают отрицательную реакцию (бесцветный раствор).
- Цератная проба заключается во взаимодействии спиртов с азотнокислым раствором гексанитратоцерата (IV) аммония, имеющим жёлтую окраску. При этом образуются переходные комплексы красного цвета, которые затем обесцвечиваются вследствие окисления спирта и перехода Ce (IV) в Ce (III).
[Ce(NO3)6]2− + ROH → [Ce(OR)(NO3)5]2− + HNO3
- Окислительная проба: при взаимодействии первичных или вторичных спиртов с реактивом Джонса, имеющим оранжевую окраску, образуются продукты окисления, а сам реактив меняет цвет на зелёный или голубой, благодаря солям восстановленного хрома (III). Важной особенностью теста является время фиксации изменения окраски — 2 секунды, по истечении которого любые дальнейшие изменения в структуре или цвете раствора не принимаются во внимание.
Количественный анализ спиртов
Для количественного анализа спиртов обычно используют методы, основанные на реакции этерификации ангидридами карбоновых кислот, например, уксусным, фталевым, а также пиромеллитовым диангидридом. Содержание спирта определяется титрованием образующейся в результате реакции кислоты гидроксидом натрия.
Другой метод анализа заключается в определении количества гидроксильных групп, способных реагировать с метилмагнийиодидом. В данном случае расчёт ведут по количеству выделившегося метана (метод Чугаева — Церевитинова).
Для гликолей применим окислительно-восстановительный метод, где в качестве окислителя используется йодная кислота. Анализ проводят по реакции образующейся йодноватой кислоты HIO3 с йодидом калия и последующим титрованием выделившегося йода тиосульфатом натрия.
RCH(OH)CH(OH)R′ + HIO4 → RCHO + R′CHO + HIO3 + H2O
5KI + HIO3 + 5CH3COOH → 3I2 + 5CH3COOK + 3H2O
2Na2S2O3 + I2 → Na2S4O6 + 2NaI
Спектральные методы анализа спиртов
Масс-спектрометрические методы анализа спиртов
Основная статья: Масс-спектрометрия
Масс-спектры алифатических спиртов имеют слабые пики молекулярного иона, а для высших и разветвлённых спиртов эти пики практически отсутствуют, поскольку в существенной степени происходит фрагментация молекулы. Фрагментация, как правило, связана с потерей молекулы воды, а также элиминированием этилена. Для длинноцепочечных спиртов преобладает отщепление воды, поэтому их масс-спектры похожи на масс-спектры алкенов. Для первичных спиртов наблюдаются пики m/z 31, для вторичных — m/z 45, 59, 73, …, для третичных — m/z 59, 73, 87, ….
ИК-спектроскопические методы анализа спиртов
Основная статья: Инфракрасная спектроскопия
ИК-спектры спиртов характеризуются двумя типами интенсивных характеристических полос поглощения:
- полосы поглощения, связанные с валентными колебаниями связи O−H: 3650—3200 см−1;
- полосы поглощения, связанные с валентными колебаниями связи С−O: 1210—1000 см−1.
Также выделяют полосы поглощения средней интенсивности, как правило, не имеющие определяющего значения: в диапазоне 1450—1250 см−1 (плоскостные деформационные колебания O−H) и 750—650 см−1 (внеплоскостные деформационные колебания O−H).
Характеристические полосы поглощения спиртов в инфракрасной области
| Типы связей и колебания | Диапазон, см−1 | Описание полосы поглощения |
|---|---|---|
| O−H, валентные колебания | ||
| ROH, неассоциированные | 3650—3580 | Узкая полоса, наблюдаемая в разбавленных растворах или парах |
| ROH•••HOR, димеры (водородная связь) | 3550—3400 | Широкая полоса, теряющая интенсивность при разбавлении |
| ROH•••HOR•••, полимеры | 3400—3200 | Широкая полоса или ряд полос |
| С−O, валентные колебания | ||
| R3COH, третичные спирты | 1210—1100 | Полосы высокой интенсивности, уменьшающейся при разбавлении |
| R2CHOH, вторичные спирты | 1125—1000 | |
| RCH2OH, первичные спирты | 1075—1000 | |
| O−H, деформационные колебания | ||
| ROH | 1450—1250 750—650 |
Широкие полосы средней интенсивности, не имеющие практического значения |
ЯМР-спектроскопические методы анализа спиртов
Основная статья: ЯМР-спектроскопия
ЯМР-спектроскопия ядер 1H широко используются для анализа спиртов, однако на величины химических сдвигов протонов гидроксильной группы (δ, м. д.) существенно влияет природа растворителя и другие внешние факторы. Для алифатических и алициклических спиртов δ составляет 0,5—3,0 (в ДМСО-d6: 4—6).
Также для изучения спиртов применяют спектроскопию на ядрах 17O. Значительная разница в сдвигах для первичных (этанол: δ 5,9 м д.), вторичных (пропанол-2: δ 39,8 м д.) и третичных спиртов (2-метилпропанол-2: δ 62,3 м д.) относительно воды H217O позволяет установить или подтвердить структуру исследуемого соединения.
Применение спиртов
Экономическое значение
Области использования спиртов многочисленны и разнообразны, особенно учитывая широчайший спектр соединений, относящихся к этому классу. Вместе с тем, с промышленной точки зрения, только небольшой ряд спиртов вносит заметный вклад в глобальную мировую экономику.
В TOP 50 за 2002 год соединений, выпускаемых химической промышленностью США, из спиртов входят только метанол (14-е место) и этиленгликоль (29-е место). В следующие 50 важнейших химических соединений, по данным за 1999 год, включены изопропиловый спирт, н-бутиловый спирт, синтетический этанол, пропиленгликоль, диэтиленгликоль, 2-этилгексанол, бутандиол-1,4, сорбит и глицерин.
Самым распространённым и используемым спиртом в мире является этанол. Его мировой объём потребления составляет около 65 млн тонн. Совокупный мировой объём потребления прочих спиртов (кроме этанола) по различным направлениям использования составляет около 70 млн тонн (по состоянию на 2009 год).
Применение в органическом синтезе
Основная доля метилового спирта используется в промышленном синтезе формальдегида методом высокотемпературного каталитического окисления метанола. Кроме того, из метанола получают трет-бутилметиловый эфир, уксусную кислоту (процесс Монсанто), N,N-диметиланилин, метиламины и хлорметан.
Из этанола в промышленности производят диэтиловый эфир (методом дегидратации при 250 °С над Al2O3), хлораль, ацетальдегид и этилацетат.
Изопропиловый спирт при каталитическом жидкофазном дегидрировании на никеле Ренея при 150 °С превращают в ацетон. Основным продуктом, получаемым из бутанола, является бутилакрилат.
Применение в качестве топлива
Основные статьи: биотопливо, биоэтанол, биобутанол
Для топливных целей в настоящий момент используются в промышленных объёмах три спирта: метанол, этанол и бутанол-1, что связано, прежде всего, с их коммерческой доступностью и возможностью массового производства из растительного сырья (кроме метанола). При этом возможно использование спиртов в виде горючего в чистом виде, в виде различных смесей с бензином или дизельным топливом, а также в качестве оксигенирующих добавок (до 10 %) с целью повышения октанового числа и снижения токсичности отработанных газов. Отдельным направлением является использование метанола для переэтерификации жиров в производстве биодизеля
Преобладающим топливным спиртом является этанол. По оценкам экспертов, на 2009 год 80—90 % всего производимого в мире этилового спирта было использовано в этих целях и составило 73,9 млрд литров (≈ 58 млн тонн).
Основными причинами, послужившими активному изучению спиртов в качестве альтернативного горючего, являются:
- рост цен на нефть и газ, а также исчерпаемость этих ресурсов в будущем;
- спирты обладают высокими эксплуатационными характеристиками, а продукты сгорания содержат меньше вредных веществ;
- спирты могут изготавливаться биохимическим методом из отходов пищевой, деревообрабатывающей и целлюлозно-бумажной промышленности, что попутно решает проблему утилизации.
Вместе с тем, массовое использование вышеуказанных спиртов в качестве моторного топлива, помимо чисто экономических причин, имеет ряд недостатков:
- метанол и этанол обладают по сравнению с бензином меньшей энергоэффективностью и, соответственно, обеспечивают больший расход;
- низкие температуры кипения спиртов могут служить причиной образования паровых пробок, что может существенно усложнить работу двигателя;
- гигроскопичность спиртов, а также их растворимость в воде может привести к резкому снижению мощности при попадании влаги в топливную систему;
- спирты обладают существенно более высокими коррозионными характеристиками по сравнению с углеводородами;
- относительно высокая скрытая теплота сгорания метанола и этанола может служить причиной проблемы при смешении этих спиртов с воздухом и дальнейшей транспортировки через впускной коллектор двигателя.
Промышленное применение спиртов
Растворители
Среди растворителей широкое распространение имеют самые разные типы спиртов: одноатомные (метанол, пропанол-2) и многоатомные (этиленгликоль, глицерин); алифатические (этанол, бутанол-1) и циклические (циклогексанол). Спирты относятся к полярным растворителям и применяются в различных отраслях промышленности. Мировой объём потребления спиртов в качестве растворителей (по данным на 2013 год) составил порядка 6,5 млн тонн .
Самым распространённым спиртом среди растворителей является этанол — его мировой объём потребления для этих целей (по данным на 2009 год) превышает 3,5 млн тонн в год. Другими популярными растворителями являются метанол и изопропанол с объёмами потребления более 1 млн тонн в год.
Использование спиртов в качестве растворителей включает в себя следующие направления:
- Технологический растворитель: экстракция и очистка натуральных продуктов (жиры и масла, смолы, воск, природные красители и ароматизаторы, альгинаты, витамины, алкалоиды, водоросли), носитель в пищевой промышленности, очистка, кристаллизация и осаждение органических химических веществ.
- Растворитель в производстве красок и покрытий: растворение синтетических полимерных материалов (лаки, смолы, клеи и т. п.), компонент цементов, красок и чернил.
- Очиститель: производство электронных компонентов, металлических поверхностей, фотоплёнок и фотобумаг, стеклоочиститель и пр., компонент жидкого мыла и моющих средств.
- Растворитель в производстве фармацевтической продукции, парфюмерии и косметики.
- Растворитель в аэрозолях (бытовых и медицинских).
Поверхностно-активные и моющие вещества
Основная статья: Поверхностно-активные вещества
Важнейшим сырьём в производстве современных поверхностно-активных веществ (ПАВ) для синтетических моющих средств являются высшие жирные спирты, из которых в зависимости от реагента получают неионогенные или анионные ПАВ.
Мировой объём использования высших жирных спиртов в производстве ПАВ в 2000 году составил 1,68 млн тонн. В 2003 году около 2,5 млн тонн ПАВ было произведено на основе высших жирных спиртов.
Полимерные материалы и их компоненты
Спирты имеют важное применение в качестве исходных мономеров для синтеза полимерных материалов методом поликонденсации. В основном, на основе спиртов синтезируют полиэфиры и полиуретаны. Важнейшими примерами таких синтетических полимеров являются полиэтилентерефталат, полибутилентерефталат, получаемые из терефталевой кислоты и этиленгликоля или бутандиола-1,4 соответственно. Поликонденсацией гликолей, глицерина или пентаэритрита с фталевым ангидридом получают алкидные смолы, которые широко используются для изготовления лаков и красок. Полиуретаны получают поликонденсацией изоцианатов с гликолями или многоатомными спиртами.
Спирты также используются для производства сложноэфирных и диэфирных пластификаторов для полимеров.
Гидравлические жидкости и смазочные материалы
Для получения негорючих гидравлических жидкостей применяют водные растворы, содержащие глицерин и этанол. В производстве тормозных жидкостей широко используют этиленгликоль и эфиры на его основе.
Многие современные смазочные материалы имеют в своём составе высшие жирные спирты и их эфиры, благодаря их низкой токсичности, высокой температуре вспышки и бесследному испарению при нагревании. Эти свойства используются для бытового применения, а также для случаев, когда эффект охлаждения поверхности более важен, чем антифрикционные свойства (например, при сверлении, пилении или другой слесарной обработке металлов).
Пестициды
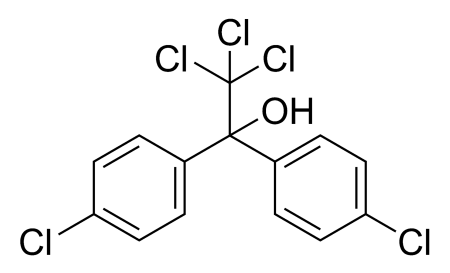
Дикопол — препарат для борьбы с клещами — ароматический спирт, аналог ДДТ.
Несмотря на то, что незамещённые предельные алифатические спирты обладают фунгицидной или гербицидной активностью, их прямое использование в качестве пестицидов не находит широкого практического применения. Одним из немногих направлений является их использование в качестве регулятора роста растений. Подобными свойствами обладают этанол, этиленгликоль и другие гликоли, некоторые высшие жирные спирты.
Галогензамещённые спирты проявляют значительно большую активность и обладают ратицидной, гербицидной и фунгицидной активностью. Так, например, препарат глифтор, представляющий смесь 1,3-дифторпропанола-2 и 1-фтор-3-хлорпропанола-2, используется для борьбы с мышевидными грызунами и сусликами.
Более высокая биологическая активность наблюдается у непредельных и ароматических спиртов. Аллиловый спирт находит применение в качестве гербицида, многие высшие непредельные спирты являются феромонами насекомых. Активными акарицидами являются некоторые ароматические бифениловые спирты: дикофол, хлорфенетол, проклонол.
Многие спирты являются полупродуктами для синтеза различных пестицидов. Например, в производстве глифосата используется метанол, кротоксифоса — α-метилбензиловый спирт, пиретроидов третьего поколения — 3-феноксибензиловый спирт.
Также спирты широко используются в качестве неводного носителя для создания товарных композиций пестицидов.
Применение спиртов в производстве потребительской продукции
Пищевая промышленность
См. также: Алкогольные напитки
Годовое потребление алкоголя на душу населения (15+) в литрах в пересчёте на чистый этанол (2004)
Основой всех алкогольных напитков является этанол, который получается при сбраживании пищевого сырья — винограда, картофеля, пшеницы и прочих крахмало- или сахаросодержащих продуктов. Кроме того, этиловый спирт используется в качестве компонента некоторых пищевых и ароматических эссенций (ароматизаторов), широко используемых в кулинарии, при выпечке кондитерских изделий, производстве шоколада, конфет, напитков, мороженого, варений, желе, джемов, конфитюров и пр. Однако этанолом список спиртов, используемых в индустрии продуктов питания, не ограничивается. Спирты можно встретить среди самых разных пищевых добавок, например, глицерин (E422) используется как — влагоудерживающий агент, растворитель, загуститель, разделитель, плёнкообразователь, средство для капсулирования. Ряд спиртов находит применение в качестве сахарозаменителей (ксилит, маннит, сорбит, эритрит), ароматизаторов (ментол), красителей (лютеин) и т. д.
Парфюмерия, косметика и бытовая химия
Спирты довольно широко используются в качестве душистых веществ для составления композиций в парфюмерно-косметической промышленности и производстве отдушек для бытовой химии и прочей потребительской продукции (гераниол, нерол, цитронеллол, ментол и др.). Помимо придания аромата, в парфюмерно-косметической продукции спирты используются и в других целях:
- антивспениватели: этанол, пропанол-1, пропанол-2, гексанол;
- антикоррозионые вещества: 2-диметиламино-2-метилпропанол-1;
- антимикробные препараты: 2,4-дихлорбензиловый спирт, (этилендиокси)диметанол;
- антиоксиданты: тиодигликоль;
- антистатики: высшие жирные спирты (C12—C16);
- гидротропные вещества: гексанол;
- маскирующие средства: бутандиол-2,3, 2-бензилгептанол-1, борнеол, 3,7-диметилнонадиен-1,6-ол-3;
- консерванты: 2-бром-2-нитропропандиол-1,3, бензиловый спирт, 1,1,1-трихлор-2-метилбутанол-2;
- охлаждающие агенты: ментол, 3-метоксипропандиол-1,2;
- пластификаторы: октандиол-1,8, 2,2-диметилпропандиол-1,3;
- противосеборейные средства: ундеканол-1;
- связующие: докозанол-1, высшие жирные спирты (C20—C22);
- смягчители: высшие жирные спирты (C9—C16); высшие гликоли (C15—C30);
- средства для завивки или распрямления волос: дитиотреитол, 3-меркаптопропандиол-1,2;
- стабилизаторы эмульсий: 7-дегидрохолестерин, 3-(октадецилокси)пропандиол-1,2, высшие жирные спирты (C9—C50);
- стабилизаторы пены: деканол-1, миристиловый спирт, цетиловый спирт, стеариловый спирт;
- увлажнители: гексантриол-1,2,6, бутандиол-1,2, бутандиол-2,3, 2-бутилоктанол-1, глицерин;
- фиксаторы волос: 2-(2-аминоэтокси)этанол;
- эмульгаторы: высшие жирные спирты (C40—C60).
Применение спиртов в медицине
Основным спиртом, применяющимся в медицинских целях, является этанол. Его используют в качестве наружного антисептического и раздражающего средства для приготовления компрессов и обтираний. Ещё более широко применяется этиловый спирт для приготовления различных настоек, разведений, экстрактов и прочих лекарственных форм.
Применение спиртов в качестве собственно лекарственных средств не столь заметно, однако многие препараты по формальному наличию гидроксильной группы можно отнести к рассматриваемому классу органических соединений (кленбутерол, хлорбутанол, маннит, эстрадиол и др.). Например, в перечне 10 наиболее важных рецептурных препаратов США в 2000 году 6-ю строчку занимает альбутерол, содержащий гидроксильную группу.
Прочие направления использования
В настоящее время трудно найти область практической деятельности человека, где бы не использовались спирты в той или иной роли. Можно выделить следующие малозначительные направления использования:
- пенообразователи для флотореагентов;
- исходные продукты для получения взрывчатых веществ, а также их компоненты (глицерин применяется для синтеза нитроглицерина, этиленгликоль — этиленгликольдинитрата, пентаэритрит — пентаэритриттетранитрата);
- исходные продукты для получения отравляющих веществ, а также компоненты для их дегазации (низшие спирты применяются для синтеза фосфорорганических боевых отравляющих веществ: табуна или диизопропилфторфосфата; моноэтаноламин и метилцеллозольв используются для дегазации).
Примечания
- Комментарии
- ↑ Показатель измерен при 22°C.
- ↑ Показатель измерен при 40°C.
- ↑ Первые три места в списке занимают серная кислота, азот и кислород соответственно. В список не входят соединения, получаемые биохимическими методами, например, этанол, получаемый из растительного сырья.
- ↑ По данным на 2009 год. Рассчитано исходя из данных потребления топливного этанола (биоэтанола) и примерных объёмов производства синтетического этанола.
- ↑ Значение рассчитано исходя из данных по объёмам мирового промышленного производства наиболее распространённых спиртов в промышленности.
- ↑ Существуют два основных технологических метода получения формальдегида из метанола:
- окисление с частичным дегидрированием (катализатор: серебро; температура: 400—720 °С):
CH3OH → HCHO + H2
2H2+ O2 → 2H2O - прямое окисление (катализатор: оксидный железо-молибденовый; температура: 300—400 °С):
2CH3OH + O2 → 2HCHO + 2H2O
- окисление с частичным дегидрированием (катализатор: серебро; температура: 400—720 °С):
- ↑ Так называемый биометанол, как и обычный метанол, производятся из синтез-газа, который в с свою очередь продуцируется из метана. При этом, при получении биометанола используется так называемый биометан, образующийся из биологических отходов в процессе жизнедеятельности специальных бактерий. Для топливных нужд может быть использован метанол любой технологии производства.
- ↑ В качестве оксигенирующих добавок помимо метанола, этанола и бутанола, могут использоваться изопропанол, пропанол, изобутанол и другие алифатические спирты.
- ↑ Разделители — вещества, облегчающие выемку продуктов из форм, противней или иных жарочных или формующих поверхностей, а также, препятствующих плотному контакту или слипанию частей продукта друг с другом.
- ↑ Плёнкообразователи — вещества, наносящиеся на поверхность пищевых продуктов с защитной целью.
|
Классы органических соединений |
|
|---|---|
| Углеводороды |
|
| Кислородсодержащие |
|
| Азотсодержащие |
|
| Серосодержащие |
|
| Фосфорсодержащие |
|
| Галогенорганические |
|
| Кремнийорганические |
|
| Элементоорганические |
|
| Другие важные классы |
|
